
根本原因就是为了避免导致 正反链混淆。
一开始,我并没弄明白,后来仔细想想也终于懂了。
如果kmer是偶数,我们会发现基因组上有些序列(如,CGCGCGCG,kmer=4)的Kmer在反向互补后得到的序列仍然是它自身!这是不能允许发生的。因为这将导致你无法区分某段序列的kmer到底是属于它自身还是说只是来自于它的互补链!!这会给解de Bruijn graph带来极大的混淆和困难!
或许你会觉得 “为什么我需要纠结于序列是不是来自互补链呢?毕竟双链DNA的正反链是严格反向互补的啊,基因组组装技术不也是把它们合并装在一起的吗?!”。你若是这样来理解其实是非常难得的,但前提却是基因组必须能够被一次性完整地(至少是非常接近完整)测出来,这时的测序深度甚至只需是1就可以了。但是你回头想想,既然都已经把基因组完整测序出来了,那还要组装干嘛呢?

并且,目前的NGS测序技术也做不到通测基因组。一般来说都是测出上百万千万亿万个小小的片段(read,长度一般是100bp-300bp)。而且,为了确保准确性,基因组都会被反复测很多层。组装时构建的kmer单位,实际上是对这些read进行的。具体的操作就是按照kmer的长度把这些read切割成更小的、存在重叠关系的片段。那么,此刻当我们构建de Bruijn graph时,如何能够保证正确地把 **同属于一条read上的Kmer连接起来,就显得极为重要了!**我们不能一会儿把A kmer正确地连到它自己所在的read,一会儿又连到它互补链的read上去!
这就是为何Kmer不能是偶数的原因了,因为只有奇数,才能保证每个kmer序列的反向互补kmer与自身也是不同的,而这个不同的真正意义就是为了避免正反链混淆。比如 :5-mer的 CGCGC,反向互补后是 GCGCG, 它们是不同的;这就不会像 4-mer,CGCG发现它反向互补后仍然是CGCG,这个时候就就会在后续连接kmer的过程中发生正负链混淆,装出一个嵌合体基因组!
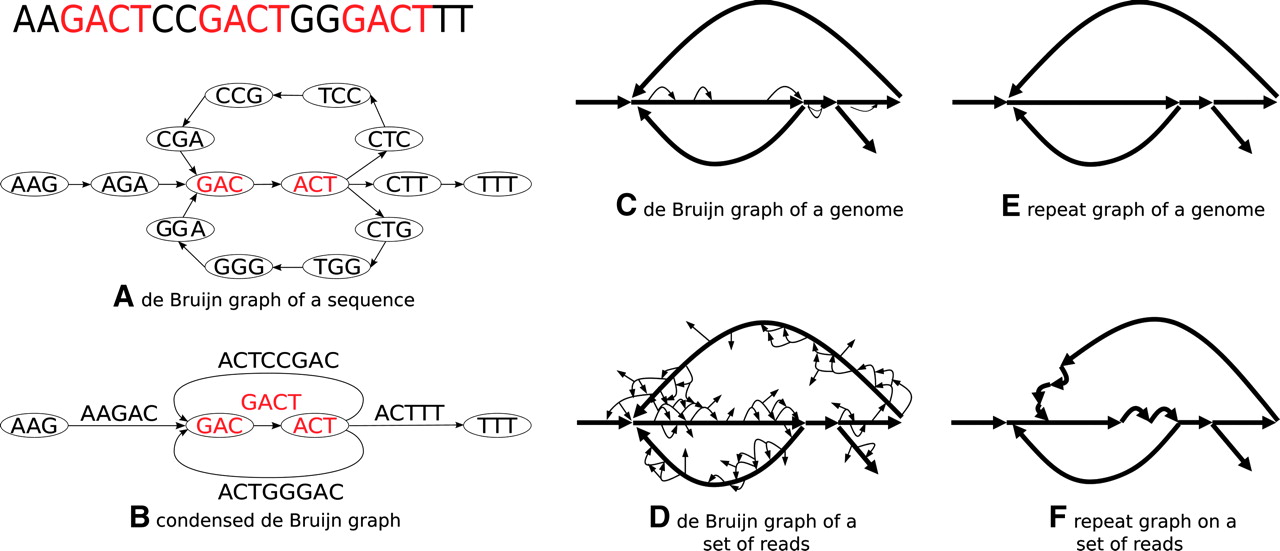
最后,放一张发表在Genome Research有关组装的图,大家可以大致感受一下这一段重复序列的组装过程。
欢迎关注我的个人公众号:helixminer(碱基矿工)
