
什么是Phasing?
Phasing,或者说Genotype Phasing,它的中文名有很多:基因定相、基因分型、单倍体分型、单倍体构建等在不同的语境下都有人说过。但不管如何,所谓Phasing就是要把一个二倍体(甚至是多倍体)基因组上的等位基因(或者杂合位点),按照其亲本正确地定位到父亲或者母亲的染色体上,最终使得所有来自同一个亲本的等位基因都能够排列在同一条染色体里面。
现在流行的NGS测序技术,都是把序列打乱混在一起测序的,测完之后,我们是无法直接区分这些序列中哪一个是父源,哪一个是母源的。我们通常都只是检测出基因组上有哪些变异,以及这些变异的碱基组成(纯合、杂合),也就是平时所说的基因型(Genotype)。只有经过Phasing,才能够实现这个区分(图1)。
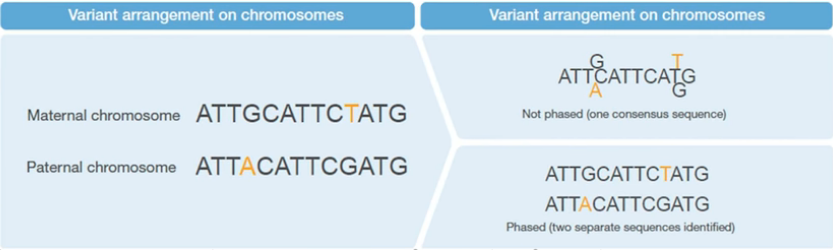
图1. 变异位点经过Phasing和不经过Phasing的示意图。右上图代表通常的Genotype,右下图代表Phasing之后的情况,实现了亲本的区分。
为什么要Phasing
**因为Phasing很重要。**Phasing的重要性可以分为两个方面。一方面, Phasing与遗传变异的功能诠释密切相关。这体现在遗传咨询师或者科学家需了解基因突变的相位后, 才能更好地判断基因突变是否会产生临床表型。比如在一个基因上发生多个Loss of function variants(LOF),通常当这些变异出于不同的单倍型时(这称为trans-configuration),即两个拷贝的姐妹基因都发生了变异, 才会导致基因表达计量(Gene expression dosage)的错误且产生危害。而当它们出于同一个单倍型时(这称为cis-configuration),因为还有一个正常拷贝的基因(作为备胎), 基因表达很可能不会发生改变也不会产生危害。
另一方面, Phasing在遗传学研究中也有诸多应用,具体如下:
- 第一、人群Phasing后形成的单倍型参考序列集(Reference panel)是基因型推断(Imputation)必须的数据材料。而基因型推断(Imputation)是基因型-表型关联分析研究中必不可少的环节。高质量的Reference Panel能提升关联分析的统计功效;
- 第二、除了Reference Panel的制造需要使用Phasing技术之外,对被研究的对象进行预先Phasing(Pre-phasing)也可以极大地提高基因型推断(Imputation)的准确性;
- 第三、使用多个位点组成的Haplotype,而不是简单的单位点基因型, 可实现群体遗传历史的推断;
- 第四、可通过Phased后的家系人群单倍型序列,估算染色体重组率、重组热点等重要遗传参数;
- 第五、Phasing可用于探测频发突变、选择信号以及基因表达的顺势调控。
Phasing说起来容易,做起来却很难
虽然Phasing理解起来并不难,但实现起来却不容易,即使在理论上也是如此。这需要相关的统计学和计算机算法技术,求解的过程往往还是一个NP问题。目前通常采用马尔科夫链蒙特卡洛算法来完成,因此,Phasing算法本身基本都是计算密集型的,做起来也比较耗时间,有时即使是在超算集群中也得跑很长时间。
Phasing的方法有哪些
Phasing的方法总结起来主要有三个:家系分型(Related individuals Phasing)、群体LD分型(LD Phasing)和物理分型(Physical Phasing)。下面我就来逐一展开对其方法进行说明。
**目前,基因定相最准确的方法是利用家系数据来实现。**具体来说,就是除了被研究的这个个体之外,同时对其父亲和母亲的基因组进行测序。有了这三个人的数据之后,就可以很容易地区分出这个样本的两个单倍体。为了便于理解,我打个比方,比如我们知道他/她的基因组某一个位置上的基因型是AB,而父亲的基因型是AA,母亲的基因型是BB,那么我们就可以清楚地知道他/她这个基因上的A是来自于父亲染色体,而B则是属于母亲染色体的,更多的具体情况可以参看下面这个示意图。
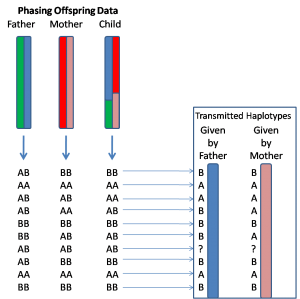
这个方法的一大优点就是定相(Phasing)的过程 非常直接、简单,不需要进行复杂的统计学计算,就可以准确地实现长距离的定相,并且还能够知道每一个基因型的亲本来源到底是什么,比如在上面的例子中,我们可以知道A和B分别属于父本和母本(如上图)。**这个亲本来源的问题对于研究或者治疗许多复杂疾病的意义是十分重大的,**比如最近发表在《Science》上的一个研究中发现,影响小孩发生孤独症(也称自闭症)的基因突变中父亲的影响更大,除此之外还有很多母源或者父源性的疾病(这里面其实还涉及到Transmitted和Non-Transmitted Chromosome的问题),这些类型的结果如果没有家系的数据是无法得出的。
对于这个方法来说,家系越庞大它的Phasing效果会越好。万一很不幸我们没能凑齐一家三口(Trio样本)仅有双样本的情况,也不用灰心,虽然效果会差一些,但还是会比没有任何族谱信息的数据要好。
家系Phasing的这个方法虽有很多难以比拟的好处,但也有一些比较明显的缺点。比如,我们为了对这个人进行定相分析,就不得不多测另外两个人的基因组。这一方面大大增加了原有的成本;另一方面则是有些人由于各种各样的原因已经难以获取其双亲的样本数据了;另外,**这个方法其实也无法完成对该个体所有变异的完全定相,**比如当碰到父、母和子/女都是杂合突变的位点时,就无法区分了。这样的位点虽然在基因组上不是最主要的,但是也大约占到了总变异位点数的13%左右,或者说有大约五分之一的杂合突变位点(注意只是占所有杂合的比例)是这种不可Phasing的状态,详细的分类情况可以参考下表:
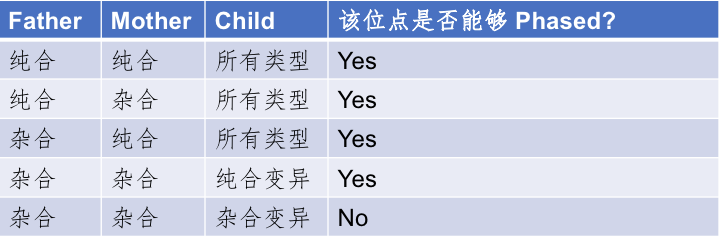
表1. 能够被Phasing和不能够被Phasing的SNPs位点分类
**LD Phasing是另外一个非常常用的基因定相方法,**它是利用群体中大量无血缘关系的个体,依据基本的连锁不平衡(Linkage disequilibrium,LD)遗传原理和相关数学模型,推断群体中每个个体的单倍体的方法,因此它也是计算量最大的一个。
我们知道人这个物种在减数分裂产生生殖细胞的过程中姐妹染色单体会发生重组,这个重组的发生率每代大约是10^-8,虽然很低,但是随着一代接一代不断地繁衍下去,经过足够长的时间之后(比如说 无穷!无穷!无穷!)。

那么从理论上来讲,来自同一祖先的两条染色就会被均匀地重组一个遍。然而,遗憾的是我们现代人还是 **Too Youg Too Simple!**从最早的证据来看现代人的共同祖先大概起源于15万-19万年前(第三次走出非洲的时间),所以至今我们也不过才经历了6,000-7,600代而已。
这么少的代数也就意味着染色体的重组其实还很有限,因此人类基因组中许多相邻的区域往往都是“黏”在一起遗传下去的,这也就是所谓的存在连锁不平衡的遗传现象,这些“黏”在一起的区块称为“连锁不平衡区块”(LD Block)。
常见变异——那些在人群中频率占比达到5%以上的变异——所存在的连锁不平衡区块(LD Block,Tajima’s D > 0.5)的长度大多集中在50Kbp-60Kbp。**并且LD区块的长度在不同的人群中是不同的,**比如,非洲人的LD区块就比欧洲人和亚洲人的更短。为什么呢?这是因为非洲人比欧洲人或者亚洲人都要更古老,他们的基因组相比于另外的两个人群发生了更多次数的重组,所以LD区块的长度就更短了。
**LD区块的存在就意味着我们可以通过构建相关的数学模型,来把这样的连锁关系求解出来。**在开展大规模的基因组研究计划时(如Hapmap、国际千人基因组、Haplotype reference consortium以及各国家的国家基因组计划),通过构建基于隐马尔可夫模型(HMM)等的Phasing算法就可以依据测序数据或者芯片数据,反推出每个个体最有可能的单倍体,完成Phasing。
目前,适合于以上两种Phasing方法(家系和LD Phasing)的最好工具是Beagle和Shapeit。这两个工具都同时包含了用于家系(Related individual Phasing)和LD Phasing的模块。并且都可以用于测序数据和芯片数据,但其中的差别在此不赘述。
回过头来想想LD Phasing方法的缺点是什么?其实通过上面的介绍,我想大家或多或少也注意到了,由于这个方法需要依据群体的信息,那么 **它所能够Phasing的精度就会受到群体的制约。**通常来说它只能针对群体中常见的变异(如频率在5%以上的变异),在这方面它的效果确实非常棒,很多基因检测公司甚至会把这个作为公司产品的买点,但对于罕见突变和个体特有的变异就不行了。虽然随着人群基数的增大,它所能够Phasing的变异范围也会随着不断增加,比如从只能Phasing 5%以上频率的变异,增大到能够Phasing 1%以上频率的变异,但说到底它还是难以实现对一个个体单倍体的完全定相。
那么,到底该怎么做才能实现完全定相呢?
正所谓,求人不如求己。由于有了以上的种种限制,于是科学家们就研发了第三类方法:Physical Phasing——「物理定相(或叫物理分型)」。它不需要家系数据,也不借助LD关系,完全依赖自身的测序数据,就可以完成基因的定相。
我们都知道在第二代或者三代测序中,一条read、一对reads或者一个clone上的每一个碱基都必定来自同一个染色体(也就是同一个单倍体)。对于每一个这样的测序片段而言,它本身就是某一个单倍体的一个“局部”,因此现在的问题就变成了要如何把这些一个一个的小”局部“连成一个整体,接出完整的单倍体,**从而实现定相,这就是Physical Phasing。**而且如果测序序列足够长(比如三代测序数据),深度足够深,那么它就能够实现个体的完全定相,而且有必要的话还可以同时把这两个单倍体的完整DNA序列组装出来,形成姐妹染色单体,这两个优点是另外两个方法难以比拟的。
长序列可以来自特殊建库,如长度约是40Kbp的Fosmid建库后的测序和组装,或者是华大测序仪的Long Fragment Read(LFR)测序技术,当然也可以是第三代测序的数据。
这也是我(矿工)在华大基因深度负责的第一个研究课题(我的博士论文也是基于该课题)。当时这一块还比较空白,第三代测序技术也还不是很成熟,当时为了获得长序列,我们采用了基于Fosmid构建大长度克隆片段然后进行二代测序并组装的方法(如下图),成果发表在2015年的《Nature Biotechnology》上,我也是共同第一作者。
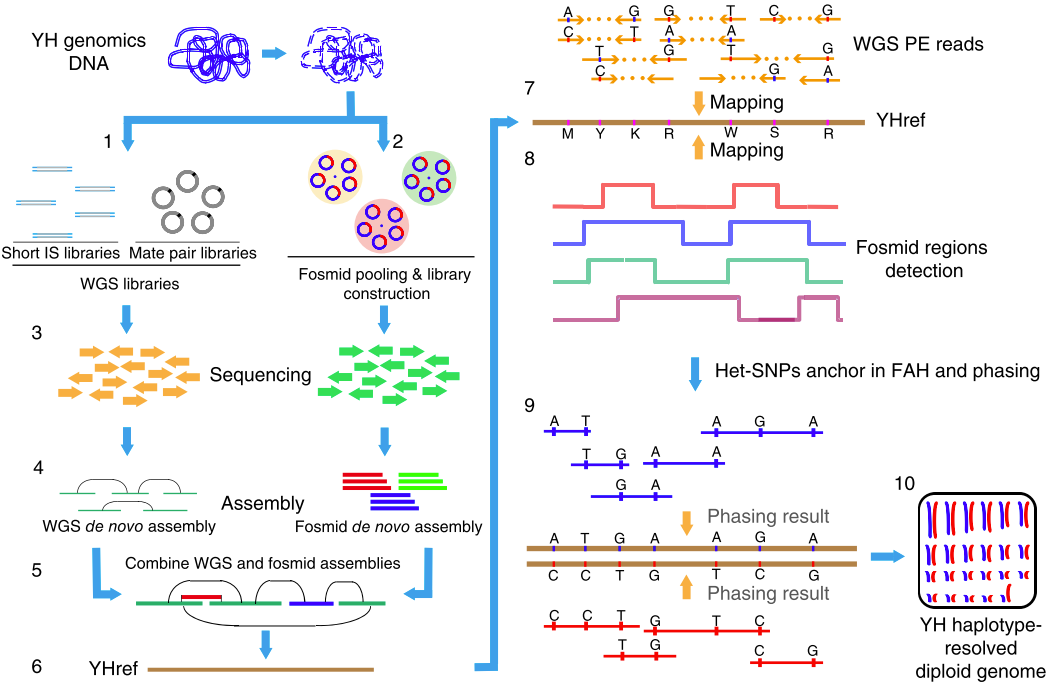
图3. 基于Fosmid和二代测序技术相结合的de novo Phasing方法
由于我当时已经为课题中的一些细节写过两篇文章,因此这里就不再展开,感兴趣的话你也可以查看本文最后的推荐阅读,这里我只介绍物理定相的基本原理。总的来说,要把局部的小片段连成一个大片段,从而实现Phasing,这个过程要做的好就需要充分借助小片段上的杂合SNPs作为区分的标记。通过每个杂合位点上各个小片段中所含碱基的异同和彼此之间的重叠关系,我们可以把绝大部分的小片段分成两类,然后通过一系列的连接、二分图构建、二分图求解和重新组装等方法,最后就可以把小片段逐步连成大片段,从而构建出单倍体了,如下图(请横着看)。
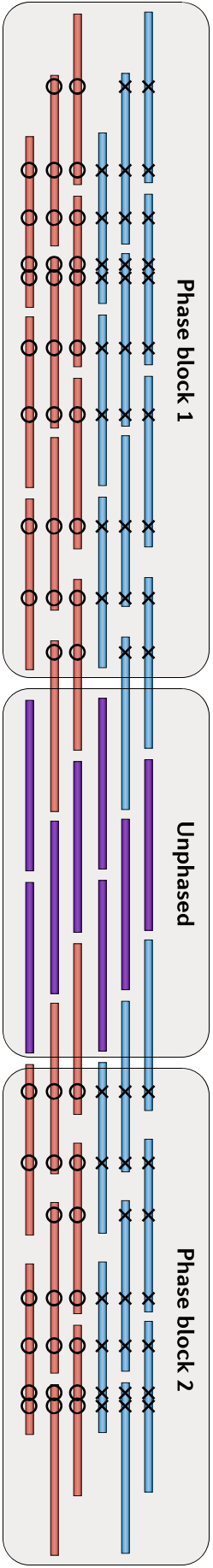
物理定相的方法,往往要求每个片段中都能包含较多的杂合SNPs位点,但由于人类基因组中杂合SNPs位点之间的距离普遍在1.5Kbp左右——还是比较长的,因此测序片段本身就要足够长,这就需要使用包括三代测序技术在内的一些测序方法,因此它的成本会比较高。我目前所知道的在Physica Phasing方面做得比较好的机构中,除了我们自己当时的小组之外,还有德国的马克普朗克研究所( Max Planck Institute)Margret教授团队和华大基因Brock Peters博士所在的研究组,他们建立了LFR的实验和信息方法。
小结
关于Phasing原理的介绍到此就告一段落了,这里在介绍LD Phasing和物理定相的时候没有从数学原理方面去展开,希望可以看起来比较通俗易懂,并且所有的Phasing算法都只对二倍体基因组比较有效,多倍体更加困难。在实际的项目中,我们还是需要根据样本的特点、测序策略和结果预期,有针对性地选择其中的一种或者多种进行组合,从而达到最有效的Phasing效果,评价Phasing效果好坏的指标有两个:
- 第一,能够被Phasing的变异位点越多越好;
- 第二,正确被Phasing的位点数占比越高越好。
推荐阅读
- 华大基因组装迄今最完整人类单倍体水平基因组的重要技术细节(上)
- 华大基因组装迄今最完整人类单倍体水平基因组的重要技术细节(下)
- GATK4.0和全基因组数据分析实践(上)
- GATK4.0和全基因组数据分析实践(下)
- 该如何自学入门生物信息学
欢迎关注我的个人公众号:helixminer(碱基矿工)

这是我的知识星球:解螺旋技术交流圈,是一个与读者朋友们的私人朋友圈,欢迎你的加入。我有9年前沿而完整的生物信息学、NGS领域的工作经历,在该领域发有多篇Nature级别的科学文章。
这是知识星球上第一个真正与基因组学和生物信息学强相关的圈子。我旨在营造一个高质量的组学知识圈和人脉圈,通过提问、彼此分享、交流经验、心得等,更好地学习生物信息学知识提升基因组数据分析和解读的能力。
在这里你可以结识到全国优秀的基因组学和生物信息学专家,同时可以分享你的经验、见解和思考,有问题也可以向我提问或者向圈里的星友们提问。
知识星球邀请链接:「解螺旋技术交流圈」