
1. RNA-Seq是否可以替代WES完成对外显子的变异检测?这不但省去用探针做外显子捕获这个步骤,代价更小;而且,它在融合基因的检测上还更有优势?
这是一个非常好的问题。我的回答是:RNA-Seq不能代替WES完成外显子的变异检测,原因如下:
(1). **转录本不是全部的外显子。**由于基因通过可变剪切出不同的转录本,实现多能性。那么,没被该转录本包括的外显子就丢失了;
(2). **转录本数据在基因上的覆盖度是极度不均匀的。**不同基因的表达量不同,有些很高,有些甚至没有。进行变异检测的时候,这种不均匀性会极大影响变异结果的有效检出。**导致很多发现的变异可能都是那些高表达但是却很可能不具备什么关注点的基因上。**如果这时你还是想获得更多的变异,那么到头来还是得花更多的钱加大测序深度;
(3). **目前对转录本数据进行变异检测,还是一个偏于补充性质的分析。**RNA-Seq的目的主要还是集中在基因表达方面,以及寻找差异表达基因和融合基因上。对于变异检测,这类数据中也肯定可以发现,但假阴一定是很高的,比如低表达的基因,甚至是在这个组织(或者样本)中不表达的基因,你就无法有效检出它基因组上的变异了。另外,由于目前的二代测序系统并不能对RNA中的U碱基进行识别,因此,RNA测序的时候需要先反转录为cDNA,这个过程会为RNA的变异检测带来一定程度的假阳结果;
(4). 变异检测范围有限。使用RNA-Seq数据很难发现除单碱基变异(SNV)之外的其他突变(比如Indel)。
2. 为什么二代测序的原始数据中会出现Read重复现象?
要搞清楚这个read重复(duplicate)的问题,我想我们需要从NGS数据的产出过程说起,具体来说如下:
- 基因组DNA提取;
- DNA随机打断,最常用的是超声打断;
- 对被打断的DNA片段进行末端修复(通常是3’加A),然后在两端加接头,选择特定长度的片段文库进行PCR扩增(通过PCR的扩增会选!择!性!地提高加上了接头的文库分子数量);
- 文库上机与测序芯片(Flowcell)上的引物结合,经过桥式PCR扩增,在芯片上形成测序所需的cluster;
- 进行SBS测序,光学信号捕获,生成序列。
我们一般认为第1步DNA提取出来的是完整的基因组,打断则是完全随机的——通常来说也确实如此。
在第3步,**PCR扩增时,同一个DNA片段会产生多个相同的拷贝,第4步测序的时候,这些来源于同!一!个!拷贝的DNA片段会结合到Fellowcell的不同位置上,生成完全相同的测序cluster,然后被测序出来,这些相同的序列就是duplicate。**这是duplicate的第一个来源,也是主要来源,称为PCR duplicates(PCR重复)。
同样,在第4步,生成测序cluster的时候,某一个cluster中的DNA序列可能搭到旁边的另一个cluster的生成位点上,又再重新长成一个相同的cluster,这也是序列duplicate的另一个来源,这个现象在Illumina HiSeq4000之后的Flowcell中会有这类Cluster duplicates,这是第二类duplicate(如下图)。
在第5步中,某些cluster在测序的时候,捕获的荧光亮点由于光波的衍射,导致形状出现重影(如同近视散光一样),导致它可能会被当成两个荧光点来处理。这也会被读出为两条完全相同的reads,这是第三类duplicate,称之为Optical duplicates(光学重复);
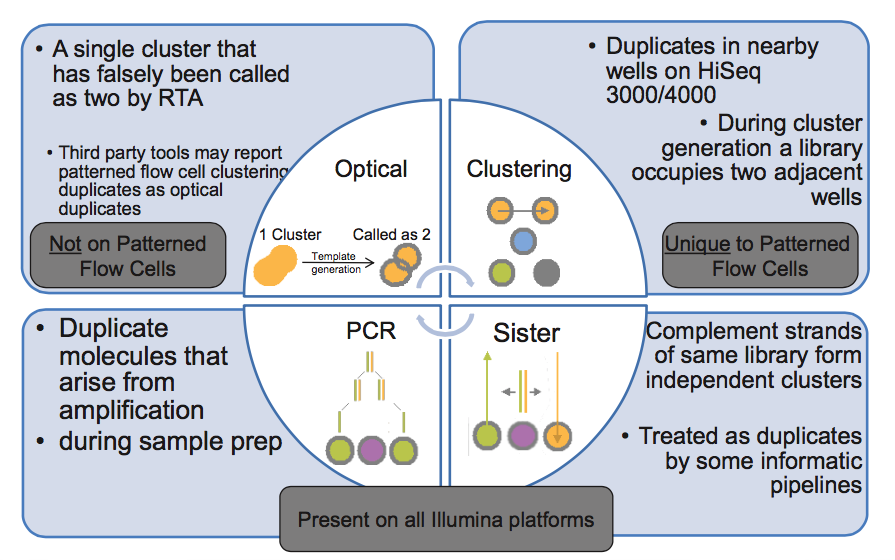
以上三种比较常见,还有第四种,**称为Sister duplicates,这是比较特殊的一个情况。**它是文库分子的两条互补链同时都与Flowcell上的引物结合分别形成了各自的cluster被测序,最后产生的这对reads是完全反向互补的。比对到参考基因组时,也分别在正负链的相同位置上,在有些分析中也会被认为是一种duplicates。
另外,据说 **NextSeq 平台上还出现过由于荧光信号捕获相机移动位置不够,导致 tile 边缘被重复拍摄,每次采样区域的边缘由于重复采样而出现了duplicates,**下图中蓝色点代表 duplicates,可以看到在tile的左右两侧明显富集。
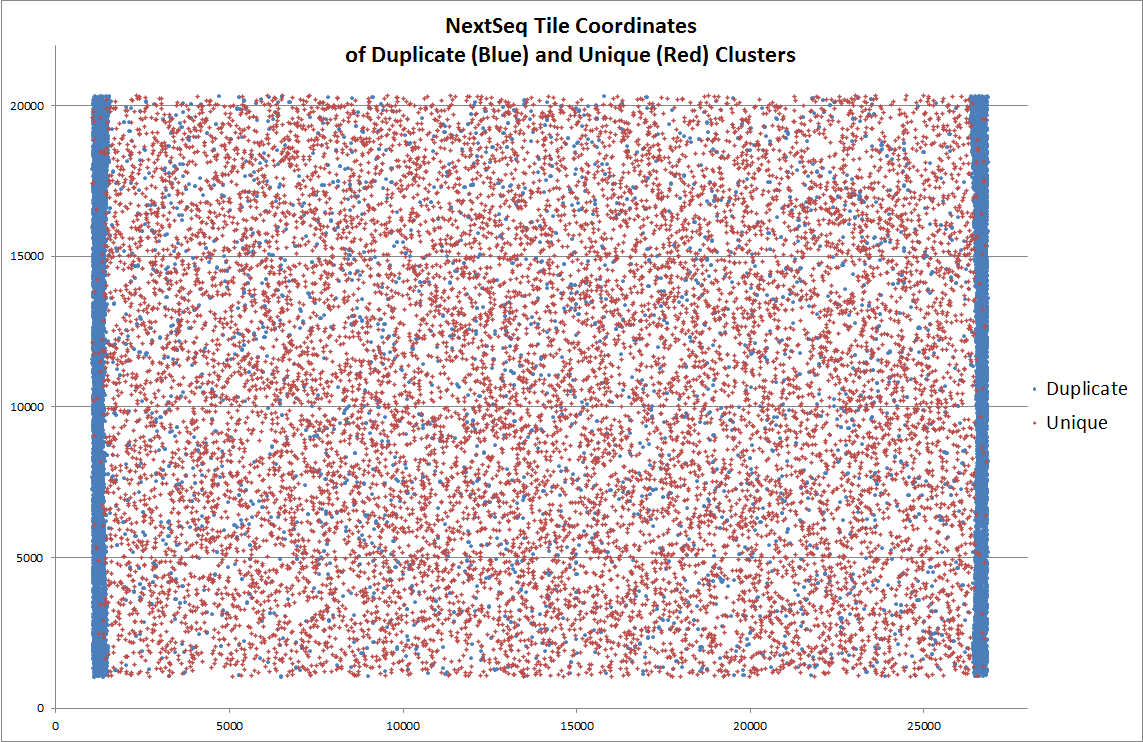
以上,除了NextSeq的情况之外,**所有这些不同类型的duplicates都各有特点。**比如,PCR duplicate的特点是随机分布于Flowcell表面;而cluster duplicates和optical duplicates 的特点是它们都来自Flowcell上位置相邻的cluster。Cluster的位置一般都会被记录在原始测序fastq文件@Sequence-id那一行中。
这些Read重复都会一定程度上导致一些碱基信号被错误地拉高或者减低,会对后续分析带来干扰,特别是在WGS和WES分析时都需要去除。如果测序过程没什么特殊问题或者原因,那么,测序数据的duplicate比例一般都在10%以下。
PCR duplicates可以通过PCR-free来避免。并且PCR本身还会带来一些其他的问题,比如扩增过程自带了一定的偏向性,这会损失一定的测序随机性,使得某些序列信息被扩大或者减小。所以,只要DNA起始量足够,那么我们就应该尽量采用PCR Free的方式来建库。
3.GATK4不能进行多线程?
![]()
当我们本地跑GATK4的时候你会发现,它竟然没有多线程的功能,这和GATK3很不同,在3中我们可以用-nt或者-nct设定多线程,但是4却没有类似的参数,这是为啥呢?
其实,关于这个问题已经有研究者问过GATK的团队。**简单来说回答是,没有!**如果要使用多核来跑流程,那么节点配置好Spark,用GATK4中的Spark功能模块(如,HaplotypeCallerSpark)就可以了,让Spark来帮你完成多线程。
**我想可能很少有人知道以前GATK3中的多线程功能的效果其实并不好,而且还容易出问题。**可能也是由于这方面的原因,GATK团队这一次在4中就干脆放弃了自己实现多线程的想法,直接使用现成的Spark来完成这个调度。另外,值得一提的是在GATK4中跑并行任务的最好做法是采用WDL和Cromwell相结合的方式。
话虽如此,**但GATK团队实际上还是留下了唯一的一个例外!那就是HaplotypeCaller中最消耗计算资源的模块——pariHMM,这个是可以本地单独多线程的!**通过--native-pair-hmm-threads这个参数来设置,它默认是4,功能有些隐蔽!
4.Ti/Tv比率能说明变异的什么特征吗?
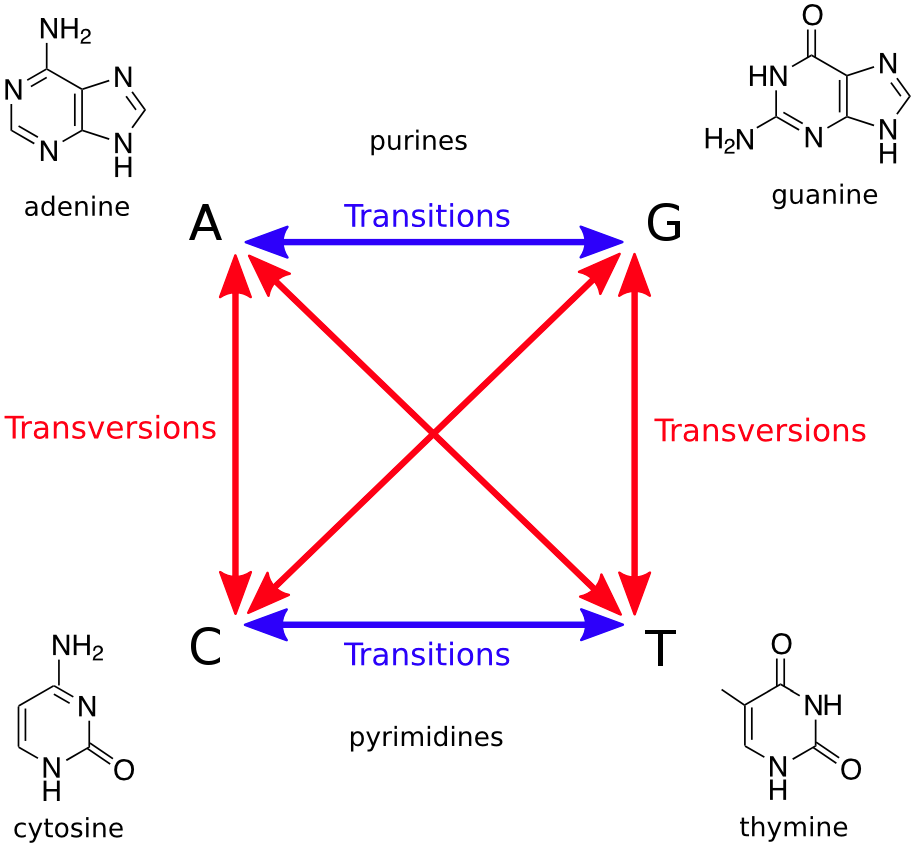
**Ti/Tv(转换和颠换的比例)的值,实际上是物种演化的过程中在基因组上留下来的序列选择标记,是对自然选择的一种反应,这儿值在物种中具有一定的稳定性。**因此,Ti/Tv的值常常会被我们作为一个评判变异的质控结果好坏的一个较为宏观的指标。
而且一般来说,在基因组上C->T的转换比较多,也就是胞嘧啶(C)容易自发转为胸腺嘧啶(T),这是因为基因组上的C在甲基化的修饰下容易发生C->T的转变。
另外,需要注意的是Ti/Tv毕竟是一个比值,它的大小和我们计算基因组上的哪一个区域有一定关系。比如对人类基因组而言,如果是全基因组区域,Ti/Tv比值一般在2.0x-2.1左右;而对于外显子区域,Ti/Tv比值是3.0左右;对于那些novel variants来说,由于还没受到明显的自然选择,它们的Ti/Tv的值会相对较低,可能会低至1.5左右。对其他区域而已,Ti/Tv范围也应该在1.5-3.0之间。
有关Ti/Tv的更多内容建议看我之前讨论变异数据应该如何质控的一篇文章。
5.最新人类参考序列的两个版本NCBI(GRCh38)和UCSC(hg38)有什么差异?
**可能大部分人都认为这两份参考序列是一致的,但实际上它们存在着细微的差别。**主要有以下四点:
(1). UCSC的hg38相比于NCBI的GRCh38缺少chrEBV(Epstein-Barr virus)序列。EBV本身不是人所有的,但由于很多细胞系在培养的过程中都需要借助EBV,因此对于许多通过细胞系测序而来的数据中(比如海拉细胞系),就会混有这个序列。在NCBI的新版本中已经加入EBV序列,但是UCSC并没有相应的更新;
(2). UCSC版本缺少decoy序列,这是参考序列中没有,但却是其他人群特有的人类序列,作为参考序列不应该缺少;另外还缺少HLA分型序列;
(3). 简并碱基的表达方式不同。NCBI的GRCh38中存在极少量的简并碱基,一共有94个,对于巨大的人类基因组序列来说,这个数字可以忽略,这些简并碱基在UCSC的版本中则用N代表;
(4). Y染色体上存在较大差别。NCBI上的GRCh38,在Y染色体上的两个PAR区域(pseudoautosomal region,伪染色体区域)用N来代替了,而UCSC并没有做类似的处理。这样会导致它们在Y染色体序列中存在明显的差异,进行序列比对时结果也会有所不同。
那么这两份我们应该用哪一份?**答案是NCBI的版本。它也是GATK bundle所使用的版本,并且NCBI对其更新和维护的节奏明显好于UCSC版本,细节也做得更好,一般我们会直接在GATK的bundle中下载。**不久前UCSC也把NCBI GRCh38直接接入到它的基因组浏览器了(上图)。
技术交流圈往期精华
欢迎关注我的个人公众号:helixminer(碱基矿工)

这是知识星球:**解螺旋技术交流圈,**是一个我与读者朋友们的私人朋友圈,欢迎你的加入。我有9年前沿而完整的生物信息学、NGS领域的工作经历,在该领域发有多篇Nature级别的科学文章。
这是知识星球上 **第一个真正与基因组学和生物信息学强相关的圈子。**我旨在营造一个高质量的组学知识圈和人脉圈,通过提问、彼此分享、交流经验、心得等,彼此更好地学习生信知识,提升基因组数据分析和解读的能力。
在这里你可以结识到全国优秀的基因组学和生物信息学专家,同时可以分享你的经验、见解和思考,有问题也可以向我提问和圈里的星友们提问。
知识星球邀请链接:「解螺旋技术交流圈」