
这源自于我2013年的一篇旧文,另外,那篇文章也已被转载得有些走样了,我决定要重新组织。虽然主体没有大的变化,但是内容在这里作了较多的翻新。对于初学者来说,如果你之前看过了我的那篇旧文,还是建议你再看一次,或许可以有新的发现,如果你从未看过,那么请不要轻易错过。
人类基因组中的变异和人类的演化、疾病风险等方面都有着密切的联系。当前二代短读长高通量测序技术(NGS),虽然能够让测序成本大大降低,但这种短读长的测序方法也给基因组的变异检测(特别是结构性变异检测)带来了不小的挑战。SNP和Indel大家应该都见得比较多了,因此在这篇文章里我将主要讨论常见结构性变异的检测方法和有关软件以及它们的一些优缺点。
变异的分类
在开始之前,有必要先梳理一下人类基因组上的变异种类,按照目前业界的看法可以分为如下三个大类:
- 单碱基变异,即单核苷酸多态性(SNP),最常见也最简单的一种基因组变异形式;
- 很短的Insertion 和 Deletion,也常被我们合并起来称为Indel。主要指在基因组某个位置上发生较短长度的线性片段插入或者删除的现象。强调线性的原因是,这里的插入和删除是有前后顺序的与下述的结构性变异不同。Indel长度通常在50bp以下,更多时候甚至是不超过10bp,这个长度范围内的序列变化可以通过Smith-Waterman 的局部比对算法来准确获得,并且也能够在目前短读长的测序数据中较好地检测出来;
- 基因组结构性变异(Structure Variantions,简称SVs),这篇文章的重点,通常就是指基因组上大长度的序列变化和位置关系变化。类型很多,包括长度在50bp以上的长片段序列插入或者删除(Big Indel)、串联重复(Tandem repeate)、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及形式更为复杂的嵌合性变异。
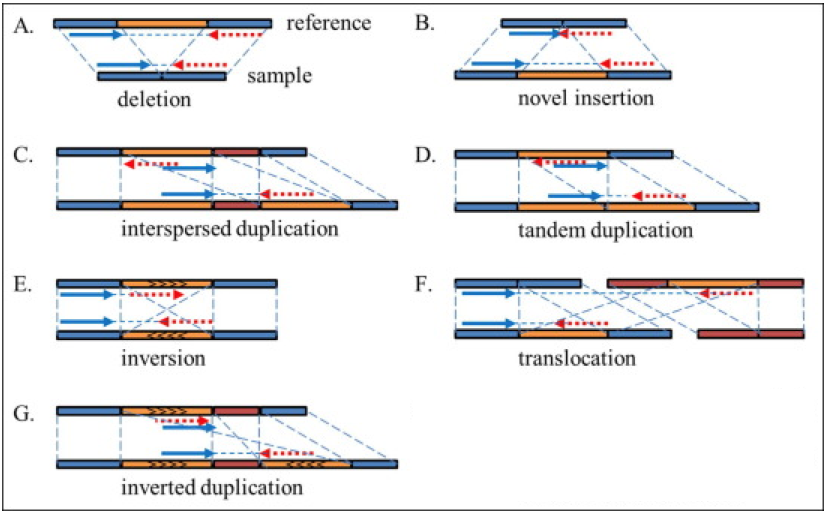
值得一提的是,研究人员对基因组的结构性变异发生兴趣,主要还是由于在研究中发现:
- SVs对基因组的影响比起SNP更大,一旦发生往往会给生命体带来重大影响,比如导致出生缺陷、癌症等;
- 有研究发现,基因组上的SVs比起SNP而言,更能代表人类群体的多样性特征;
- 稀有且相同的一些结构性变异往往和疾病(包括癌症)的发生相互关联甚至还是其直接的致病诱因。比如,《我不是药神》电影中提到的慢粒白血病,它就和基因组的结构性变异直接相关。它是由于细胞中的9号染色体长臂与22号染色体长臂相互易位,导致ABL基因和BCR基因融合,形成了一个会导致ABL异常表达的小型染色体(称为费城染色体)发生的。这是一个典型的结构性变异致癌的例子。
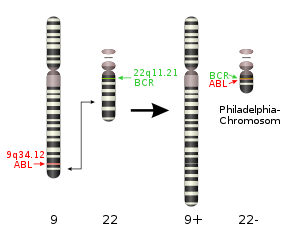
图2. 9号染色体和22号染色体长臂发生易位,形成"费城染色体",常导致慢性粒细胞白血病。没错!也就是《我不是药神》中可以用”格列卫“治疗的一种白血病
类似的例子还有很多。不过应该注意的地方是,大多数的结构性变异并不直接与疾病的发生相关联,很多也不一定会致病,它还与周围环境的响应或者其他的一些表型多态性有关。
**一个东西一旦引起多方关注,很快就会红起来,SVs也是这样的。**近年来,在NGS技术快速发展的大背景下,SNP和Indel这类比较容易发现的变异的检测算法已经做得七七八八了,大家就开始选择新的未解问题——研究热点,所以在这些情况的加持下,人类基因组上的结构性变异就逐渐被全面而又集中地研究了起来,相应的变异检测工具和算法也越来越多,如下附图3,在omictools上就有超过130个与SVs有关的工具。
除了测序的方法之外,一些专用的基因芯片也可以用于检测一部分结构性变异,好处是成本低,但是局限大,而且芯片技术实际上只对大片段的序列删除这类变异比较灵敏,无法做到全面,同时做不到单碱基精度的断点检测。
工具虽然不少,但其实归纳起来,基于NGS数据的变异检测算法并不多。总的来说主要有以下4种不同的策略和方法,分别是:
- Read Pair,一般称为Pair-End Mapping,简称RP或者PEM;
- Split Read,简称SR;
- Read Depth,简称RD,也有人将其称为RC——Read Count的意思,它与Read Depth是同一回事,顾名思义都是利用read覆盖情况来检测变异的方法;
- 序列从头组装(de novo Assembly, 简称AS)的方法。
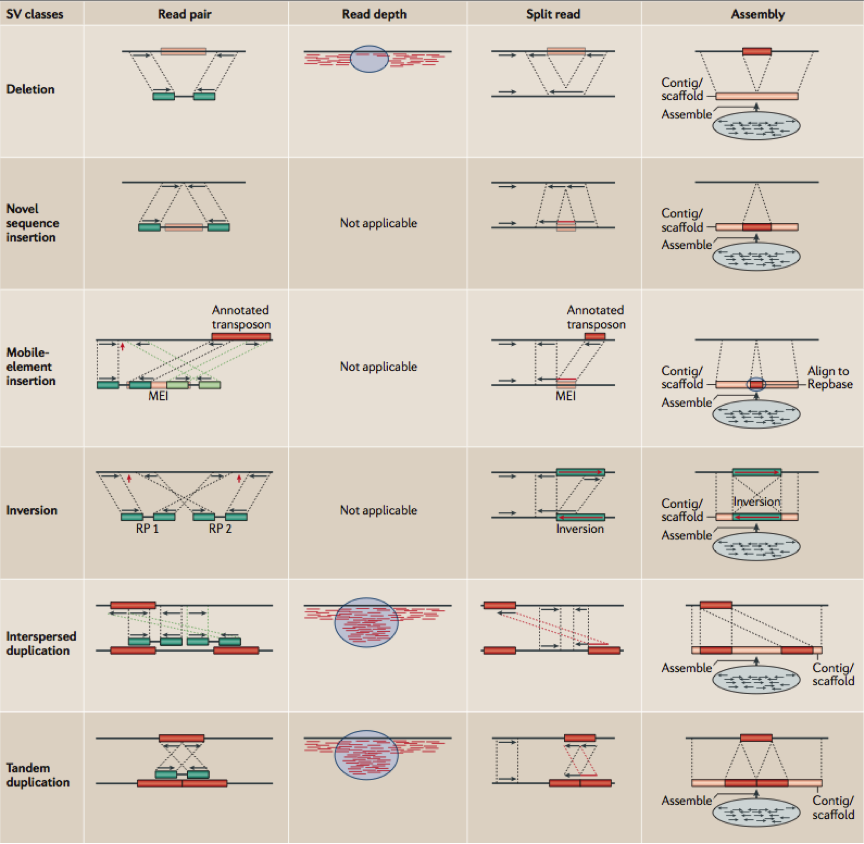
图4. 四种类不同的算法进行结构性变异的检测分类,行是结构性变异类型,列是算法
接下来我将对这四种不同的方法以及它们各自的特点逐一进行展开介绍。但是,具体要用什么软件,参数要怎么设置,命令要怎么跑,这写操作性的内容我就不在这篇文章中展开了,大家可以看其参考文档(如果它的参考文档没有写明白,那么多半说明这个软件做的不怎么样,慎用)。这篇文章的目的是想让大家对这些SVs方法有一个宏观上的认识,让你知道原来基因组上结构性变异检测是这样的。
基于Read Pair(RP),也就是Pair-end Mapping(PEM)的方法

这是RP方法的一个主要框架,用的比较多。它的思想是这样的:我们知道PE测序的两条read(通常称为read1和read2),它们原本就是来自于同一个序列片段的(如下图,对此不清楚的同学也可以查看这一篇文章)。

因此,Read1和Read2之间存在着客观的物理关联(它们就是在一起的),而且它们之间的距离——图中这个淡蓝色序列片段(通常称为插入片段)的长度,称为插入片段长度(Insert size)。一般来说我们是无法直接获得每一对read1和read2之间真实的插入片段长度的,但通过序列比对,计算它们彼此之间比对位置上的距离却可以间接获得这个长度——BAM文件的第9列记录的就是这个值(这里是BAM的格式和关于使用Pysam处理BAM文件的文章[https://mp.weixin.qq.com/s/c0O5qHBnybZNKCERLzYbJQ]中我还举了提取插入片段长度的例子),如下图:
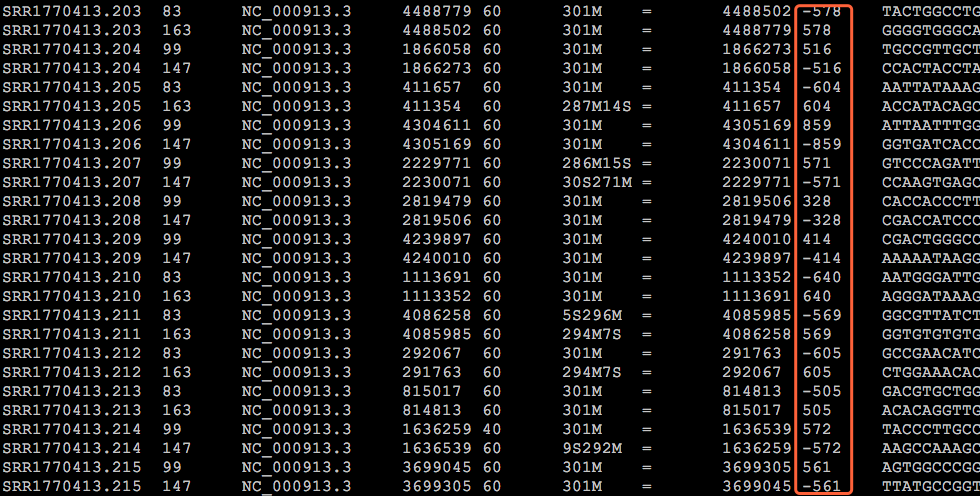
图7. 在BAM文件中可以方便地得到Insert size信息
**这个插入片段长度的分布是RP方法进行变异检测的一个关键信息。**我们知道,在测序之前需要先用超声或者酶切的办法把原始的DNA序列进行打断处理,然后再挑选某一个长度(比如说500bp)的DNA片段来上机测序。在这个过程中,虽然我们都希望挑选的序列是一样长的,但这肯定是不可能的。**事实上,我们得到的片段长度通常都会围绕某个期望值(比如我们这里的500bp)做上下波动。**如果我们把这些按照比对位置计算获得的“插入片段长度”提取出来做个分布图,会看到它的样子通常如下:
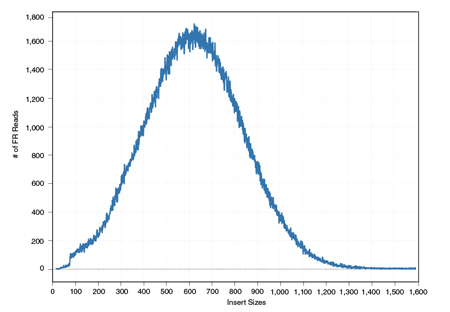
没错!**如无意外你见到的一般都是类似这样的一个钟形线——正态分布,**理论上它也应该是一个正态分布。而且由于基因组上存在变异的序列毕竟是少数,所以,这个分布本身就能够反映“插入片段长度”在绝大多数情况下的真实分布情形,那么对于那些小部分不能反映真实情况(偏离分布中心的片段长度)的是什么呢?是隐含的变异信号!
**因为,如果插入片段长度有异常,它实际上包含的意思是,组成read1和read2的这个序列片段和参考基因组相比存在着序列上的变异。**举个例子,如果我们发现它这个计算出来的插入片段长度与正态分布的中心相比大了200bp(假设这个200bp已经大于3个标准差了),那么就意味着参考基因组比read1和read2所在的片段要长200bp,通过类似这样的方式,我们就可以发现read1和read2所在的序列片段相比与参考基因组而言发生了200bp的删除(Deletion)。RP除了可以利用异常插入片段长度的信息进行线性变异(特指Deletion和Insertion)的发现之外,**通过比对read1和read2之间的序列位置关系,还能够发现更多非线性的序列变异。**比如,序列倒置(Inversion),因为,按照PE的测序原理,**read1和read2与参考基因组相比对,正好是一正一负,要么是read1比上正链,read2比上负链,要么是反过来,**而且read1和read2都应处于同一个染色体上,**如果不是这种现象,那么就很可能是序列的非线性结构性变异所致,**比如前者是序列倒置(Inversion),后者是序列易位(Translocation)等。
总的来说,RP通过利用比对距离、read1和read2之间的位置关系这两个重要信息实现了基因组上多种结构性变异的检测。
利用这样的原理,RP方法理论上能够检测到的变异类型可以包含如下6种:
- 序列删除(Deletion)
- 序列插入(Dnsertion)
- 序列倒置(Inversion)
- 染色体内部和染色体之间的易位(intra- and inter-chromosome translocation)
- 序列串联重复/倍增,也就是常说的Tandem duplications
- 序列在基因组上的散在重复(Interspersed duplications)
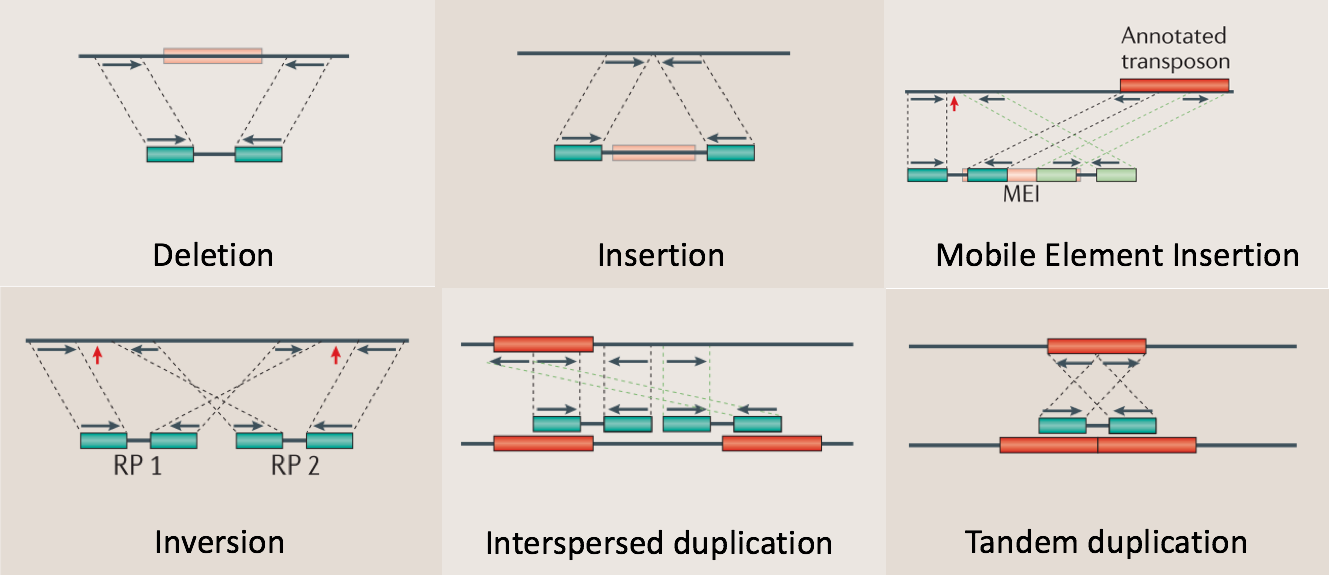
图9. 适合RP策略检测的6类结构性变异,蓝绿色的小块代表的是测序Read
应该说检测的范围还是比较广的,但对于RP存在缺陷的地方需要指出:
- 第一,对于Deletion的检测,由于要求插入片段长度的变化要具有统计意义上的显著性,所以它所能检测到的片段长度就会受插入片段长度的标准差(SD)所影响。简单来说,**就是越大的序列删除越偏离正常的长度中心,才越容易被检测到。RP对于大长度Deletion(通常是大于1kbp)比较敏感,准确性也高,**而50bp-200bp的这个范围内的变异,由于基本还处于2倍标准差以内,在统计检验上它的变化就显得不那么显著,所以这通常就成了它的一个检测 暗区。
- 第二,它所能检测的Insertion序列,长度无法超过插入片段的长度。因为,如果这个Insertion序列很长——举个极端的例子——整个插入片段都是Insertion序列,那么你会发现read1和read2根本就不会比对上基因组,它在基因组上一点信号都没有,你甚至都不知道有这个序列的存在。另外,Insertion的检测精度也同时受限于插入片段长度的标准差。
Delly和Breakdancer都是应用RP方法的SVs检测软件,应该也是当前用得比较广的软件,可能相比于Breakdancer,Delly用得还要更多一些。此外,Delly不仅在进行结构性变异检测的时候应用了RP,它还同时使用了SR的方法。
Split Read(分裂read,简称SR)
SR这个方法,起源于Sanger长片段测序数据变异检测的场景,而且如果单条序列足够长(>400bp),那么它还可以有效地检测出MEI(Mobile Element Insertion)。NGS迅速发展起来之后,测序序列变成了短序列,不过SR同样被应用过来了。
插播一句,媒体常常把NGS被翻译为“下一代”测序技术,实际上这是一种不明所以的翻译,说是二代测序技术也就算了,这个“下一代”是啥?是还没来吗?现在都到三代了。还不如说是测序的2G和3G时代。所以,我还是更喜欢用NGS这三个字母。
在短序列数据中 **SR算法的核心也是对非正常PE比对数据的利用。**这里有必要和上文RP(Read-Pair)中提到的非正常PE比对做个区分。RP中的非正常比对,通常是read1和read2在距离或者位置关系上存在着不正常的情形,而它的一对PE read都是能够“无伤”地进行比对的;但SR一般是指这两条PE的read,有一条能够正常比对上参考基因组,但是另一条却不行的情形。这个时候,**比对软件(比如BWA)会尝试把这条没能够正常比上基因组的read在插入片段长度的波动范围内,使用更加宽松的Smith-Waterman局部比对方法,尝试搜索这条read最终可能比对得上的位置。**如果这条read有一部分能够比上,那么BWA会对其进行软切除——soft-clip(CIGAR序列中包含S的那些read,图9中也有这类比对情况),标记能够比上的子序列(但不能比上的序列还是留在原read中,这也是软切除的含义)。但这个过程有时候可能不会太顺利,甚至会发生多次切除再比对的情况,所以,你会看到一条read有时候竟然有很多个Soft-clip的比对结果(这个不是指多比对的情况,而是多次切除的情况),而发生这些情况的read就是SR方法的用武之地。并且soft-clip保留原序列的方式对于后续应用SR很重要,因为,它们往往不会只是依赖原有BWA的比对结果,而是会对这条read进行重新局部比对(如果没有保留那么信息的丢失就会导致大量的假阴),比如Pindel就是这么做的,下文详述。
除了Pindel,Delly也是应用SR方法进行变异检测的软件,不过Pindel在早期千人基因组计划中用的最多,Delly是后起之秀,当然现在有代表性的工具还有不少,比如lumpy、SVseq2等。
图10,展示了SR方法所能检测到的变异类型以及它是如何用这些Split reads的信号来进行结构性变异检测的,其中的红色线段就是被split出来并重新比对的序列。可以看出在不同的变异类型中,这些split的信号是不同的,而且它们和RP相比也很不同,SR的read通常都会被“撕裂”出来,而RP则是“无伤”完整的read,大家注意对比这个区别。
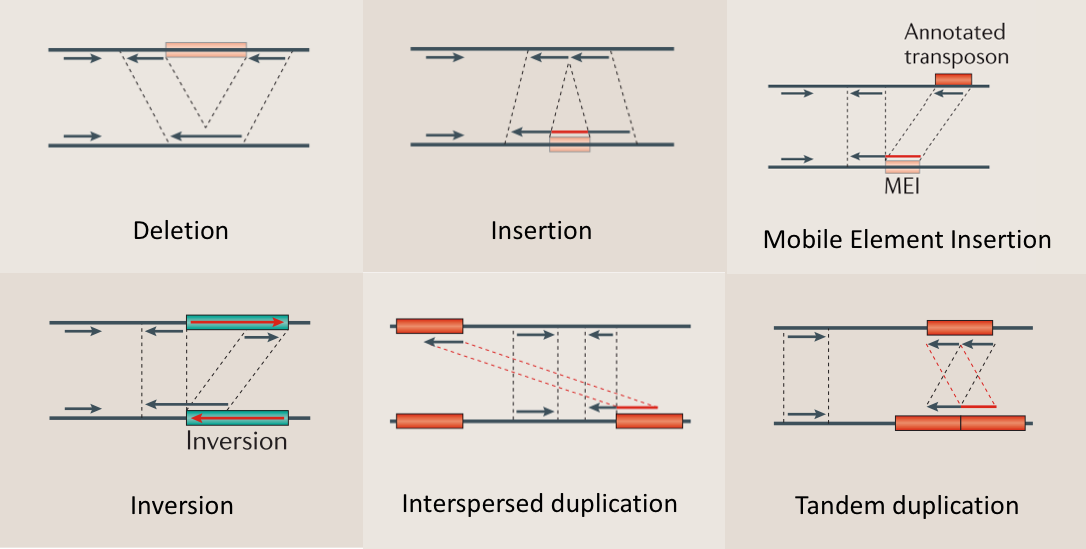
以Pindel为例子,再说一下其利用SR进行变异检测的思想原理。
首先,在获得了单端唯一比对到基因组上的PE read之后,Pindel会将不能正常比上的那条read切开成2或者3小段。然后,分别按照用户设置的最大deletion长度重新进行比对,并获得最终的比对位置和比对方向,而断点位置的确定就根据soft-clipped的结果来获得。
Pindel理论上能够检测所有长度范围内的Deletion以及小片段的Insertion(<50bp),Inversion,Tandem duplication和一些large insertion也都能够找到。
SR和RP有一个共同之处,**同样会利用比对的方向去判定相关的变异,它俩虽然方式和方法有区别,但所检测到的变异是存在重叠和互补关系的。**SR的一个优势在于,它所检测到的SVs断点能精确到单个碱基,但是也和大多数的RP方法一样,无法解决复杂结构性变异的情形。
而且 **对于SR来说,它要求测序的read要更长才能体现它的优势,**比如上文我提到的,如果有400bp以上的长度,那么一些MEI或者Alu序列的变异都能检测到,read太短,许多变异都会不可避免地被漏掉,它的检测功效在基因组的重复区域也会比较差。
Read Depth
Read Depth(有时也叫Read Count)简称RD,是目前解决基因组拷贝数变异检测(Copy number variantion,简称CNV)的主要方法。拷贝数变异通常包括,序列丢失和序列重复或倍增两个大类,在肿瘤基因组数据分析中用的比较多。其实CNV实质上是序列Deletion或Duplication,是可以归类于Deletion和Insertion这个大的分类的,只是由于它的发生有着其独特的特点,而且往往还比较长,所以也就习惯了独立区分。
RD的原理基于read覆盖深度。全基因组测序(WGS)得到的覆盖深度呈现出来的是一个泊松分布——因为基因组上任意一个位点被测到的几率都是很低的——是一个小概率事件,在很大量的测序read条件下,其覆盖就会呈现一个泊松分布,如下图。
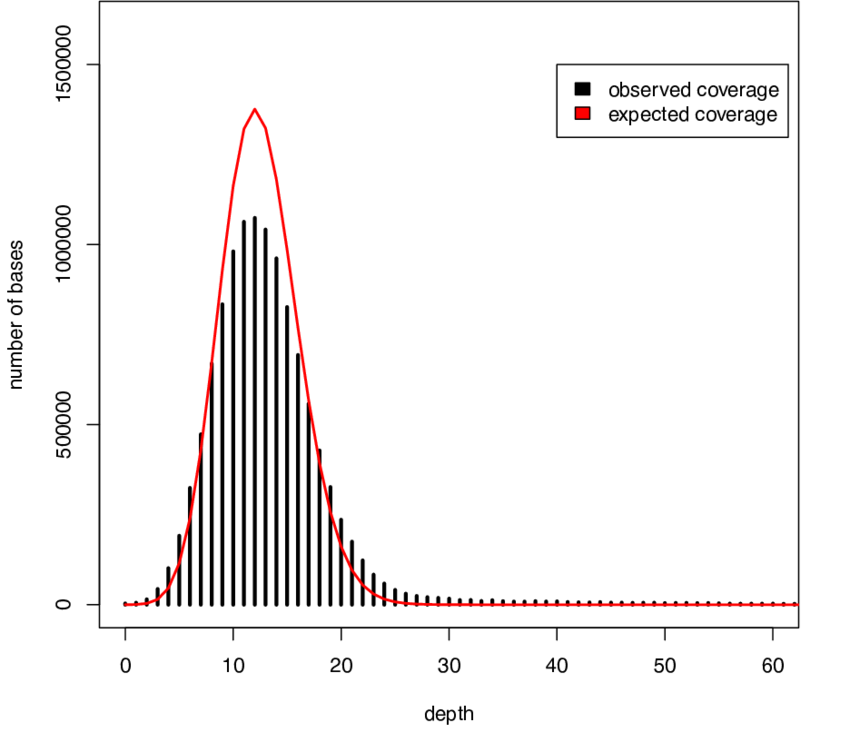
目前有两种利用Read depth信息检测CNV的策略。一种是,**通过检测样本在参考基因组上read的深度分布情况来发现CNV,**这类适用于单样本,也是用的比较多的一个方法;另一种则是通过识别并比较两个样本在基因组上存在丢失和重复倍增的区域,以此来获得彼此相对的CNV,适用于case-control模型的样本,或者肿瘤样本Somatic CNV的发现,这有点像CGH芯片的方法。
CNVnator使用的是第一种策略,同时也广泛地被用于检测大的CNV,当然还有很多冷门的软件,这里就不再列举了;CNV-seq使用的则是第二种策略。
基于其原理,RD的方法可以很好地检测一些大的Deletion或者Duplication事件,但是对于小的变异事件就无能为力了(如图9所示)。
基于 de novo assembly
**其实从上面看下来,SVs检测最大的难点实际上是read太短导致的。**就因为read太短,我们不能够在比对的时候横跨基因组重复区域;就因为read太短,很多大的Insertion序列根本就没能够看到信息;就因为read太短,比对才那么纠结,我们才需要用各种数学模型来 猜测这个变异到底应该是什么等等。
那么既然如此,就去想办法加长read的长度啊!办法有两个:三代长read测序和序列从头组装(de novo assembly)。
目前不太好评价三代测序和从头组装哪个更好,它们各有优势也各有各的问题。比如,三代测序错误率高和对Indel错误的引入会带来比较大的纠错成本,而组装则对数据量有比较高的要求,而且装重复区域也有困难等。
**但从理论上来讲,三代测序和de novo assembly 的方法应该要算是基因组结构性变异检测上最有效的方法,它们都能够检测所有类型的结构性变异。**以组装为例子,就目前来说,它是大长度 Insertion和很多复杂结构性变异的最好检测方法(这是所有基于短序列比对方法的盲区)。
我们在丹麦人国家基因组项目中用的就是这方法,其中的基于长序列的SVs检测算法也是我主要开发的,效果还不错。我们发了三篇文章(第一篇Pilot1在《Nature Communication》上,方法学一篇在《GigaScience》上,以及最后的主文章在《Nature》上),应该说是做出了有史以来最完整的人群基因组结构性变异图谱,填补了以前很多地方上的空白(图13),这个项目从2015年到目前为止加上我们和丹麦三所高校应该已经发了10来篇文章了。
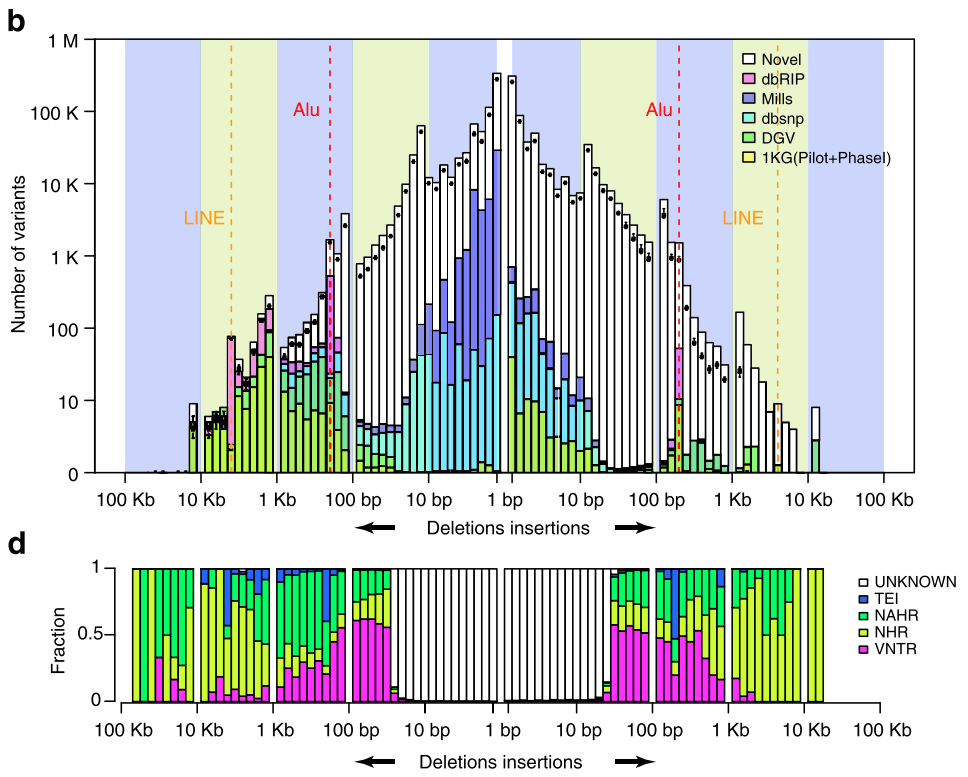
图12. 柱子的白色部分是其它检测算法所发现不了的SVs暗区,但我们在利用组装的方法在丹麦人基因组中解决了
但就像我前面说到的,序列从头组装要装的好,还是比较棘手的,对高等植物和脊椎动物来说,也是如此。最主要的原因在于,这些物种基因组上所存在的重复性序列和序列的杂合会严重影响组装的质量,除去资金成本,这也在很大程度上阻碍了利用组装的方法在基因组上进行变异检测的应用。
最后,结构性变异检测的方法这么多,我该如何选择
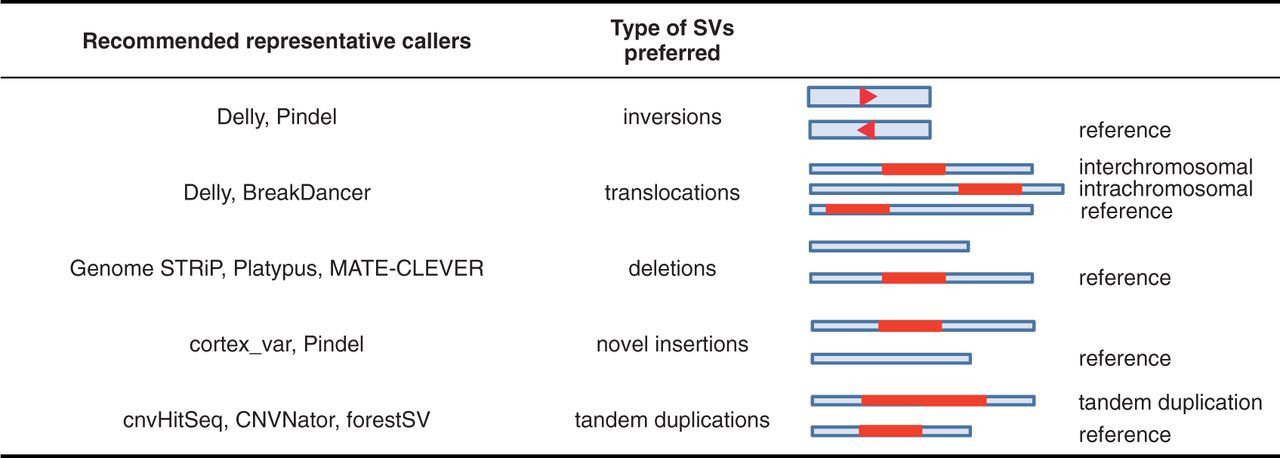 上面这个表列出了目前主流工具所适合的SVs,可以根据实际情况进行选择。
上面这个表列出了目前主流工具所适合的SVs,可以根据实际情况进行选择。
其实通过上面对四种不同SVs检测策略的比较也可以发现,小长度范围内的变异以及较长的deletion,问题不大,但对于大多数的Insertion和更复杂的结构性变异情况,当前的检测软件基本都没法还解决。**Assembly应是当前全面获得基因组上各种变异的最好方法,但是目前的局限却也发生在Assembly本身,若是基因组没能装得好,后面的变异检测就更是无从说起。**从目前的情况看,de novo assembly的方法并不能很快进入实际的应用。**因此,暂且不提assembly,其余的三种策略都各有各的优势,从目前的结果看,并没有哪一款软件能够一次性地将基因组上的各种不同情况变异类型都获得。因此就目前短reads高通量测序技术来说,最合适的方案应是结合多个不同的策略,将结果合并在一起,这样可以最大限度地将FN和FP降低。**Speedseq和HugeSeq应该是这方面的一个典范。
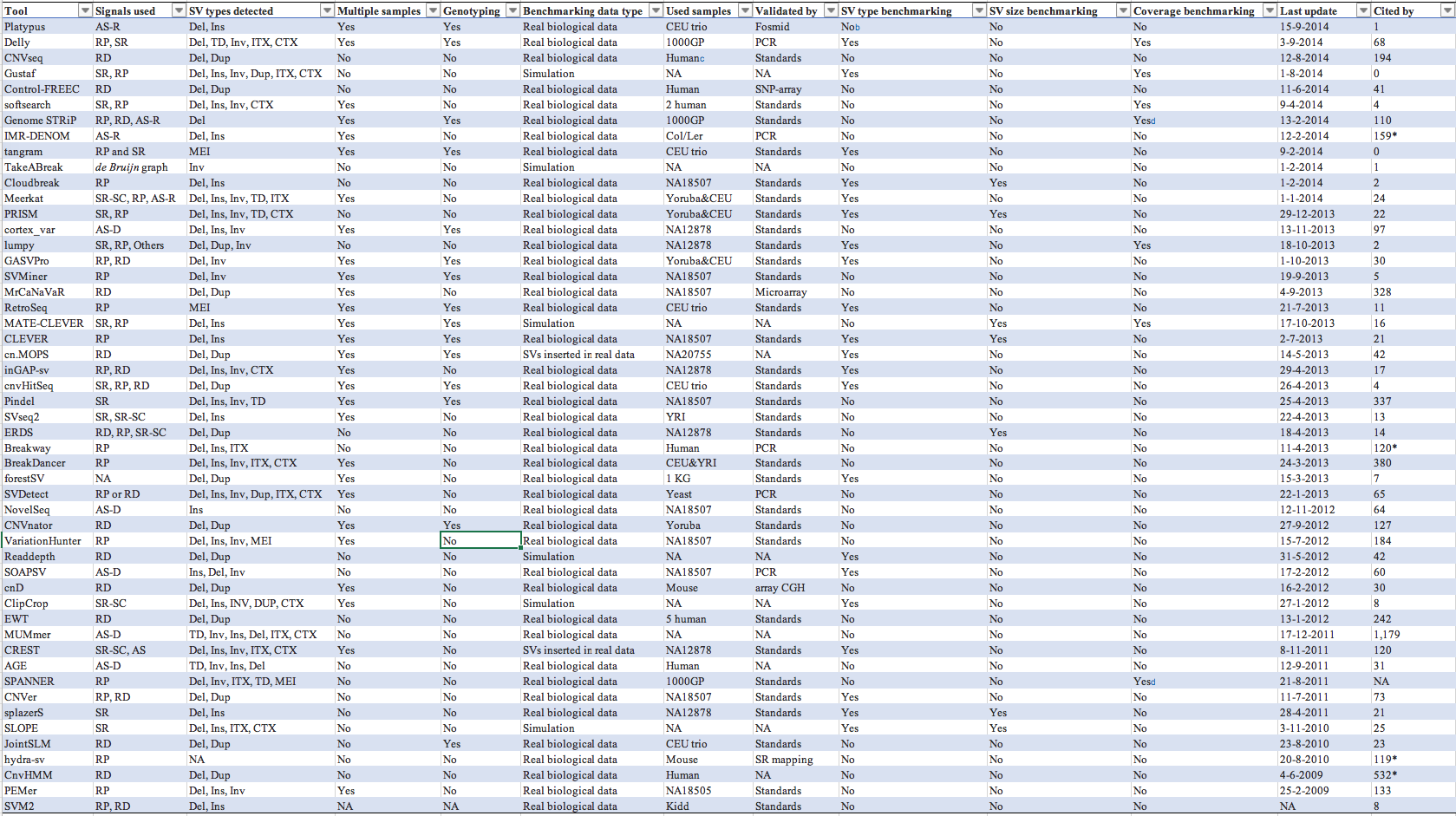
文末,再来一个各类SVs检测方法大比拼的大表供大家参考,很有价值不要错过哦,它来自于这篇文章,如下:
参考文献
- DePristo, M. a et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics43, 491–8.
- Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nature reviews. Genetics12, 363–76.
- Mills, R. E. et al. Mapping copy number variation by population-scale genome sequencing. Nature470, 59–65.
- Africa, W. A map of human genome variation from population-scale sequencing. Nature467, 1061–73.
- Korbel, J. O. et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome biology10, R23.
- Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxford, England)25, 2865–71.
你还可以看
本文首发于我的个人公众号:helixminer(碱基矿工)


这是知识星球:『解螺旋技术交流圈』,是一个我与读者朋友们的私人朋友圈。我有9年前沿而完整的生物信息学、NGS领域的工作经历,在该领域发有多篇Nature级别的科学文章,我也希望借助这个知识星球把自己的一些微薄经验分享给更多对组学感兴趣的伙伴们。
自从星球正式运行以来,已经过去了7个月,星球的成员已经超过 260人了。所分享的 主题超过了500个,回答的问题超过了140个,精华70个。我在知识星球上留下的文字估计也 已经超过10万字,加上大家的就更多了,相信接下来星球的内容一定还会不断丰富。
这是知识星球上 第一个真正与基因组学和生物信息学强相关的圈子。我希望能够借此营造一个高质量的组学知识圈和人脉圈,通过提问、彼此分享、交流经验、心得等,彼此更好地学习生信知识,提升基因组数据分析和解读的能力。
在这里你可以结识到全国优秀的基因组学和生物信息学专家,同时可以分享你的经验、见解和思考,有问题也可以向我提问和圈里的星友们提问。
知识星球邀请链接:「解螺旋技术交流圈」