
2018年10月4日,十一黄金周还没过,朋友圈摄影大赛正在如火如荼地进行着,我们这篇文章《Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and history in Chinese populations》就在 Cell 主刊上正式发表了,这也是华大内部百万中国人基因组学项目的第一期成果。
 图 1. Cell 文章. 第一作者:刘斯洋、黄树嘉、陈芳、赵立见和袁玉英
图 1. Cell 文章. 第一作者:刘斯洋、黄树嘉、陈芳、赵立见和袁玉英
项目所用的数据来自非侵入式无创产前基因检测(以下简称: NIPT),这是一个全基因组超低深度测序数据。这一期一共包含了141,431个样本,但每一例样本的测序深度都非常低,只有 0.06x-0.1x,相当于每个人只随机测了其整个基因组上 6%-10% 的区域。这和普通的WGS(10x-30x)或者以前的低深度(>1x)测序数据差异巨大,所以我们只能用 “超低” 这个词来形容。
虽然从 Cell 文章的结果来看,我们主要完成了:群体遗传学、多种表型的 GWAS 和血浆病毒谱这三大方面的内容。但其实项目刚开始时我们所设想的路线比这个要更杂一些,如图2。最后是因为有些研究点的结果并不如预期,或者是辅助数据实在不好找,计算资源也没那么多,便搁置或者放弃了。
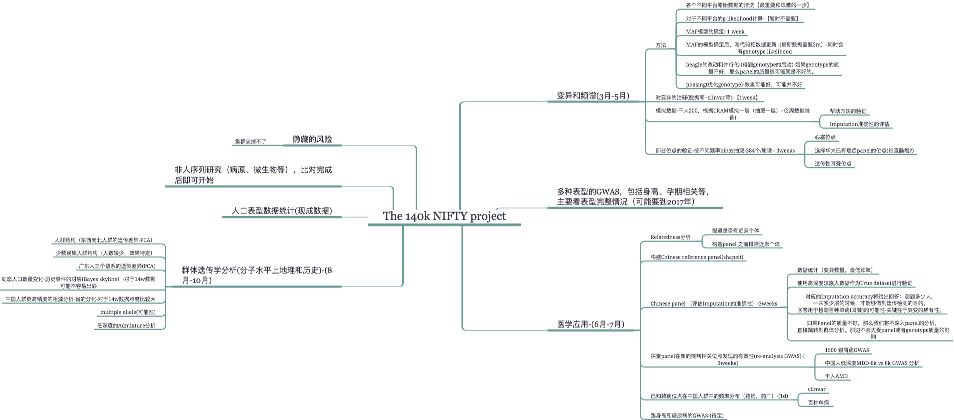 图 2. 2016年一开始的计划
图 2. 2016年一开始的计划
精准医学和基因诊断离不开大规模的人群遗传基线数据
 图 3. 全球主要的大规模基因组学计划
图 3. 全球主要的大规模基因组学计划
自 2001 年人类基因组计划完成之后的近二十年时间里,测序技术发展快速,成本不断下降。其带来的一个结果就是,世界上一些有远见的发达国家和地区——主要是美国和英国,就不断由政府、研究机构或者企业推出一系列基因组学“大手笔”项目(图3),这里面比较有名的有:美国百万老兵基因组学项目、英国10万人基因组计划(GenomicsEngland,国庆期间听说已经启动了第二期达到500万人!)、英国UK biobank等。这些项目的成果有些我们已经看到了,它们带来的好处不仅是推动了本国的基因组学研究和精准医学的发展,我觉得更重要的还在于它们逐渐树立起了强大的国际影响力和领域话语权。
中国是世界上第一大人口国和第二大经济体,拥有着至少56个不同的民族,遗传资源很丰富,想必也很独特,但一直以来,由我们主导的中国人基因组学研究成果却比较有限。
不过这个情况正在发生改变,咱们国家也开始推动大人群项目了,包括:哈工大“中国十万人基因组计划”,当时我还对这个计划做了一点评价,感兴趣的小伙伴可以移步到《我如何看,今日央视宣布我国启动“中国10万人基因组计划”》、金力教授发起的泰州人群队列项目(这也是一个10万级别的人群队列项目)等。另外,也有远在牛津大学的陈铮鸣教授发起的CKB项目——含有约10万人的基因芯片数据,还有就是企业机构发起的大人群项目,这其中就包括我们华大。
但由于很多项目才刚开始不久,因此,现在被广泛使用作为中国人代表的基因组数据集依然仅有“国际千人基因组计划”中的三百余个样本。
不得不说,在国家级人群基因组学研究以及该领域的影响力方面,我们还是略有落后。精准医学计划也搞了好几年了,但起色甚微,其中很重要的一个方面是缺少大规模的地区性人群遗传基线数据研究。当然,这其中原因有很多,包括:大型项目的设计、大规模样本的采集相当困难、测序成本也还不是真正的“白菜价”,一个上万人规模的基因组学项目依然需要不小的经费支持。比如英国的GenomicsEngland,他们要测10万人(截至今年9月已经测了8.7万人),前期的项目投资就高达7,800万英镑,折合人民币是7个亿,而它二期的投入则是一个500万人的组学计划!
但我们国家却也很幸运,你如果回过头来看,会惊讶地发现我们国家在基因技术的应用方面走得很快。特别是近年来,NIPT技术的发展和推广,其实已经让中国成为了地球上拥有最多可分析基因数据资源的国家之一,那些数据已经产生,它不需要你重新去测序!所以,这也就成了我们这个项目的一个契机,看看是否可以以这类数据为突破口,完成大规模的组学研究,如果能够成功或许可以开拓一个新的组学大数据研究思路。
万分之一中国人遗传学大数据研究
目前全球的NIPT测序数据估计已经超过了1200万例,其中大约70%的检测数据发生于我国。由于没有找到官方的报道,所以这个数字是我根据去年的情况推算的。
华大在2016年第一次达到一百万 NIPT 检测量,现在已经超过 350 万了。从数字上看, 也有着很多的可能,但在高兴之前,我们发现由于NIPT数据在测序策略方面的特殊性,导致目前大多数的组学数据分析方法都不能够适用!所以你会发现以前发表的很多NIPT文章基本都只停留在医学检测层面,但这恰恰是我们的机会!既然有难度,我们只要突破了就很可能成为第一。
NIPT数据特殊的“恶劣”条件: 1)NIPT数据来自血浆游离DNA(cfDNA)这和普通WGS不同,cfDNA大部分来自凋亡的白细胞,另一个是肝脏细胞,长度峰值在166bp左右; 2)测序深度只有0.06x-0.1x,这比起前两年COVERGA的数据还要低一个到两个数量级; 3)含有一定比例的胎儿DNA,是一个特殊的混合数据。read也非常短。我们项目中的数据为35bp和49bp,其中35bp占据大部分。虽然这对于分析染色体的非整倍体,如T21,T18和T13完全足够,但对于精细化的科学研究却困难重重,这也是这个课题刚开始时碰到的第一个挑战。
所以,当2016年3月斯洋和我一起来做这个项目的时候,我们就意识到这里面大有可为(那个时候我们刚完成丹麦人国家基因组项目),所以就一起很快制定了项目的路线(图2)。斯洋更是在完成路线设计之后就迅速写出了文章初稿,并搭建了主体数学模型。接着就是密集的讨论,在随后很短的时间内我们就完善了模型,不久之后我也把算法实现了,这个在很大程度上影响了项目的整体执行进度和路线。
那时,其实我们也知道 ,留给我们的时间并不多——科学只认第一不认第二,一旦落后心血也就白费了。所以后续的分析也基本是在急行军,并没有什么休息不休息的,只想快点做出来,一旦想/找到一个能够解决问题的方法,立刻就上。最后到2017年12月的时候就基本完成了。
这个项目在开始的时候,我们就做了分期,第一期只用14万例NIPT测序数据——全部都是早期NIPT检测的超低深度全基因组数据*——这个数据的选择也经过了一番思考和伦理审查,**最后留下的数量是141,431例**。这个数据在 2015年 的时候也发过一篇医学检测的文章,而且**很巧的是这个数量大约为全国人口的万分之一**,样本数据也广泛分布于全国各地(覆盖中国31个省级行政单位和36个少数民族),再加上NIPT检测的样本来源天然就有随机性,所以不难看出这将是一组非常有代表性的中国人群体数据(图4)。
这个数据前期已经完成了清洗、质控、比对和比较等方面的工作,这是团队中张勇、徐惠欣和李胜康所完成,并且做出了很多的努力和尝试!
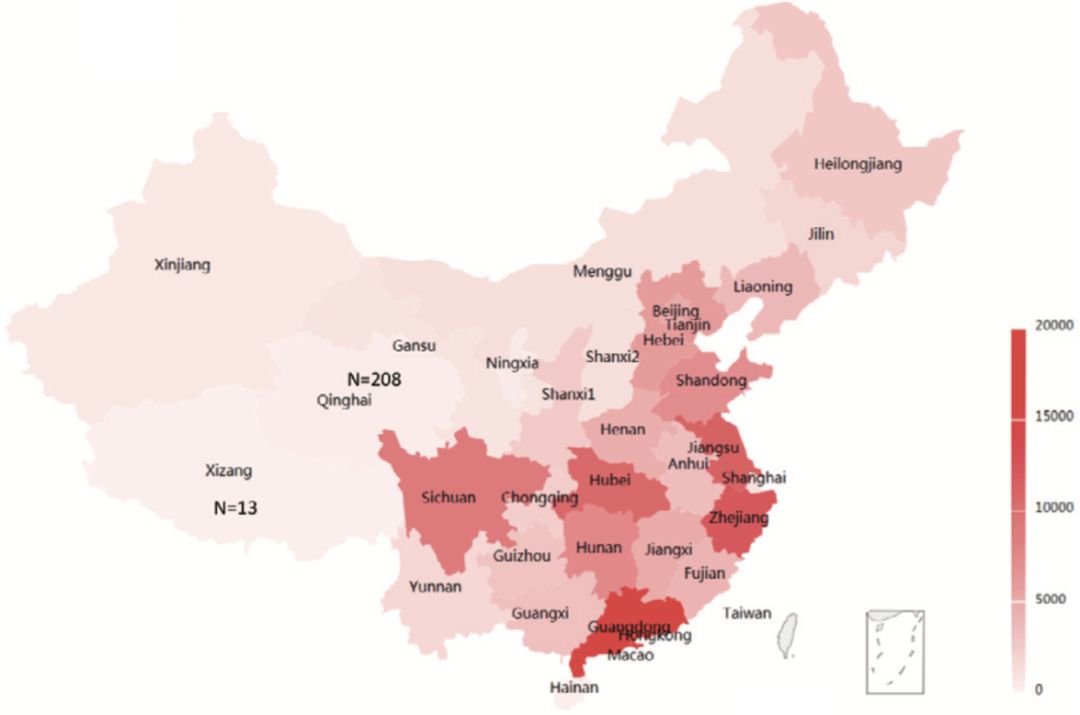 图 4. 14万人的分布
图 4. 14万人的分布
项目过程中我们解决了不少方法学上的难题,包括开发了适用于此类数据的分析方法和算法。但更大的目标是,要借助这个项目建立一套基于超低深度测序数据进行组学大数据研究的方法和策略,不断发掘 NIPT 数据的价值,并为第二期做准备。这其中涉及的技术和方法,我们在下一篇文章中讨论,本文先说做了什么。
中国人群体的生物大发现
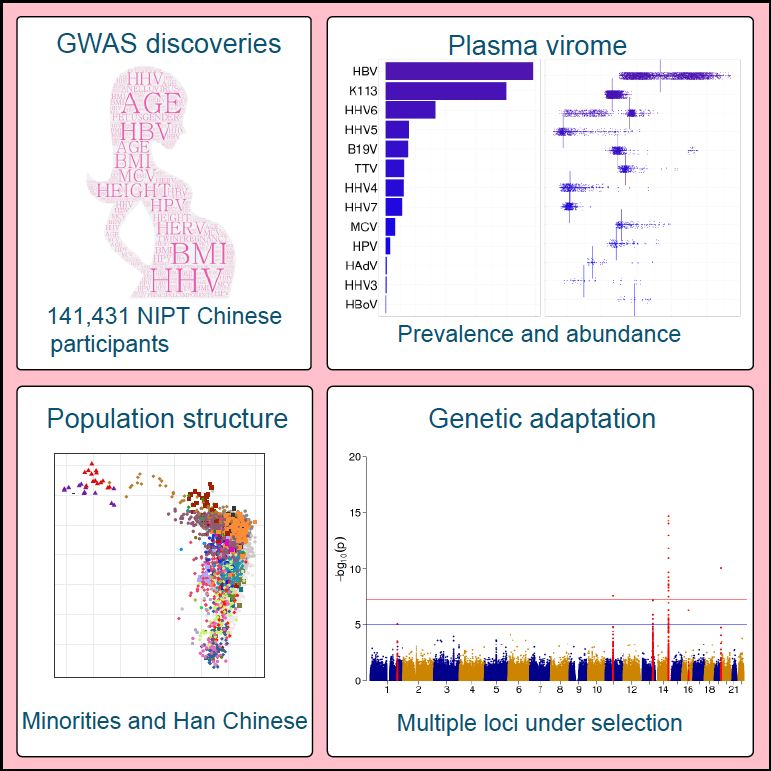 图5. 研究摘要
图5. 研究摘要
这个图是这一期Cell文章的研究摘要,下文逐个展开说明。
群体变异检测
首先是群体变异检测(注意是群体,而不是个体),这是一定要得到的一个结果,但这里是无法用GATK这类工具来完成的,深度太低,样本太多,也不适合采用个体genotype的思路。所以我重新构建并开发了一套变异检测算法(名字叫:BaseVar),完成了这块内容。最后用14万人的数据,一共得到了904万个高质量的单碱基多态性位点(原始的SNP结果则超过3400万),其中包含约20万个新发现的多态性位点。同时还把它们做成了数据库,我们把它命名为:Chinese Millionome Database(简称:CMDB,已经可以注册使用了),从名字你也可以看出来,我们的目标是百万人!
不过,此刻你可能会诧异,为什么14万人才得到9百万左右的SNP,是不是有点少了?其实你如果了解我上述提到的“恶劣”条件,就不难想象了,即使把所有人加起来在基因组上每个位置平均也差不多只有1万条read覆盖,再考虑测序错误、比对错误、变异的置信度判断等,能够质控过关的也差不多是这个数了。即便如此,这也已经是目前登顶学术期刊的最大规模中国人群基因频率数据集了。
接下来是这个研究中比较出彩的部分。
群体遗传学揭开人群遗传差异
 苗族同胞土鸡火锅 | 图源: Getty
苗族同胞土鸡火锅 | 图源: Getty
这一次我们在群体遗传学方面做了不少工作。首先是通过构造人群距离矩阵,分析遗传距离的变化和基因流方向,很清晰地得到了汉族和少数民族群体各自的遗传结构特点。比如,认识到汉族人原来是一个遗传连续体,有着明显的南北方向遗传分层,却没有明显的东西差异——我这里姑且称为“东西同宗”现象(文章没有这个词)。这个很可能与我国存在大规模汉族人口西迁有关,而且由于分离时间不长,西部汉人与东部汉人的差异还没拉开。相比之下少数民族则有着较多更为精细的结构。
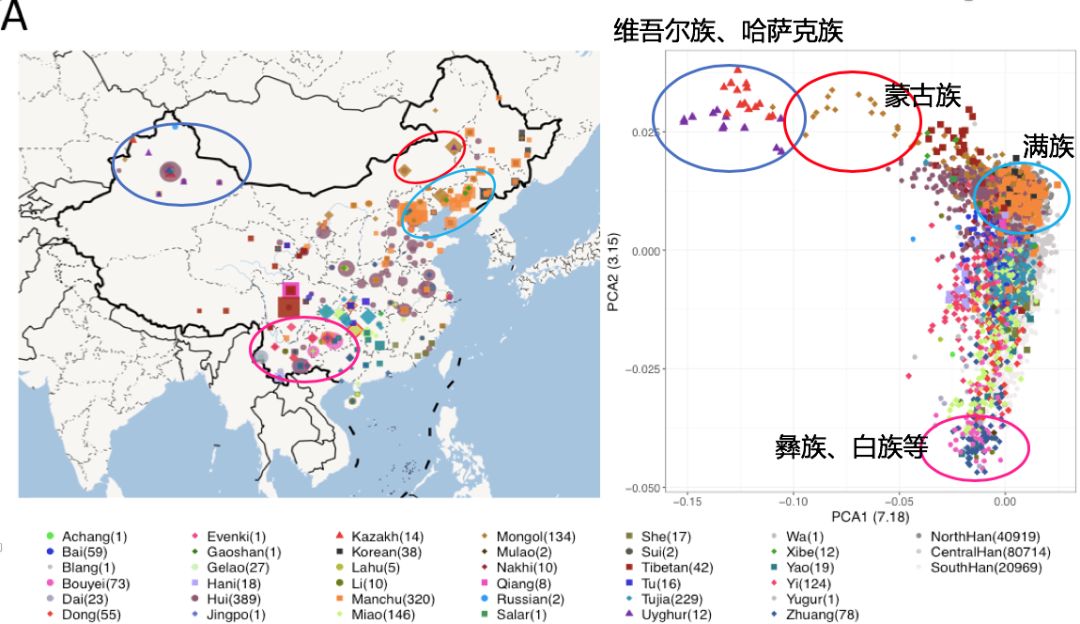 图6. 少数民族遗传结构,左图是少数民族聚集的地方,右图是少数民族群体的遗传结构
图6. 少数民族遗传结构,左图是少数民族聚集的地方,右图是少数民族群体的遗传结构
比如图6,我们以汉族为背景(灰色)作比较。其中与汉族差别最大的主要是维吾尔族、哈萨克族、蒙古族,结构边界也很清晰。而中部的回族、西南的彝族、白族和南方的壮族、布依族也与汉族不同,只是边界不是很清晰。满族则和北方汉族没太大区别,这个可能是因为历史上彼此就生活在一起,而且清朝时满汉一家亲,基因交流甚是频繁。。。
基因流
这是群体遗传学中的一个重要内容,搞清楚自家的关系之后,我们也很好奇咱们中国人和国外其它群体之间的遗传关系到底是怎么样的。
鉴于数据量上的优势,我们一次性比较了各省与欧洲、南亚以及东亚人群的基因交流程度(通俗来说就是通婚程度)。结果有了一个比较有趣的发现,如图7,左边的地图是各省所有中国人(包括少数民族同胞)和欧洲人基因交流程度的情况,颜色越红,代表携带的欧洲血统越高。左图这部分的结果并不让人意外,甚至还很直观——基本是在地理(包括文化习俗)上越接近欧洲的要越高,所以你可以看到新疆、青海、甘肃和内蒙古地区的欧洲人成分相比于其它地区都要高得多,而且呈现了一个从西北到东南的递减规律。
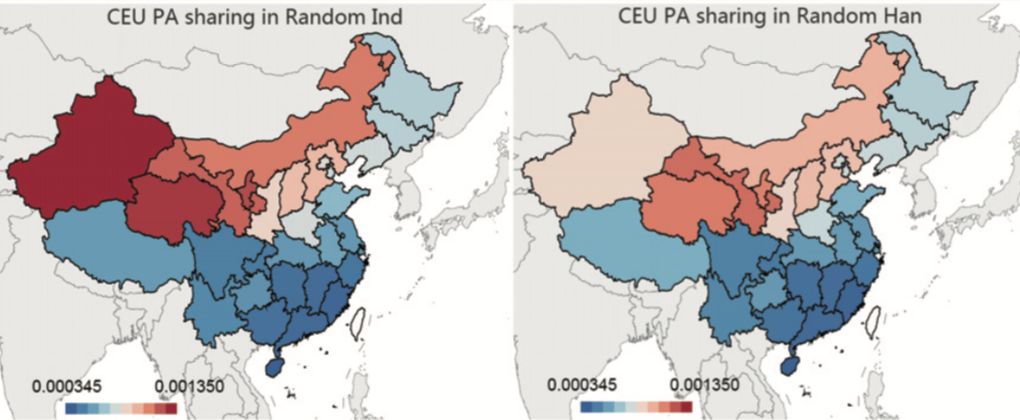 图7. 各省包括少数民族在内的全体中国人(左)和各省汉族人(右)所含有的欧洲人成分分布
图7. 各省包括少数民族在内的全体中国人(左)和各省汉族人(右)所含有的欧洲人成分分布
但是当我们把其中的少数民族抽掉,只看各省的汉族人与欧洲人的基因混合情况时(右图),却有了一个较为反常的发现,那就是生活于甘肃省、青海省和宁夏的汉族人所含有的欧洲人成分明显高于其它省份,甚至高于在新疆地区生活的汉族人,为全国平均水平的1.7倍,达到0.115%!这有点奇怪,为什么就这里那么突出呢?

经过一番分析之后,我们注意到这个地区恰巧是古代中国丝绸之路的必经之地——河西走廊的所在之处(或者附近)!这里原本就是古代中西方人群汇集和交流的地方,很可能是这个原因导致该地的汉族人有着较高的欧洲人血统。
然后我们又分析了与印度人的基因交流程度,呈现的是从西南到东北的递减规律,这个没有太多意外,所以这里就不重复了。
南北方6大遗传差异基因
我们还找到了6个在纬度方向上受到强烈自然选择的基因(图8)。其中3个与机体免疫功能有关,1个与脂肪代谢有关,1个与骨密度及精神类疾病(如,躁郁症)有关以及1个与耳垢干湿、大汗腺分泌、体味有关。从左上角曼哈顿图可以看到这些差别信号很显著,很可能是饮食、气候、病原微生物等环境因素对我们中国人的演化所起到的选择作用所致。
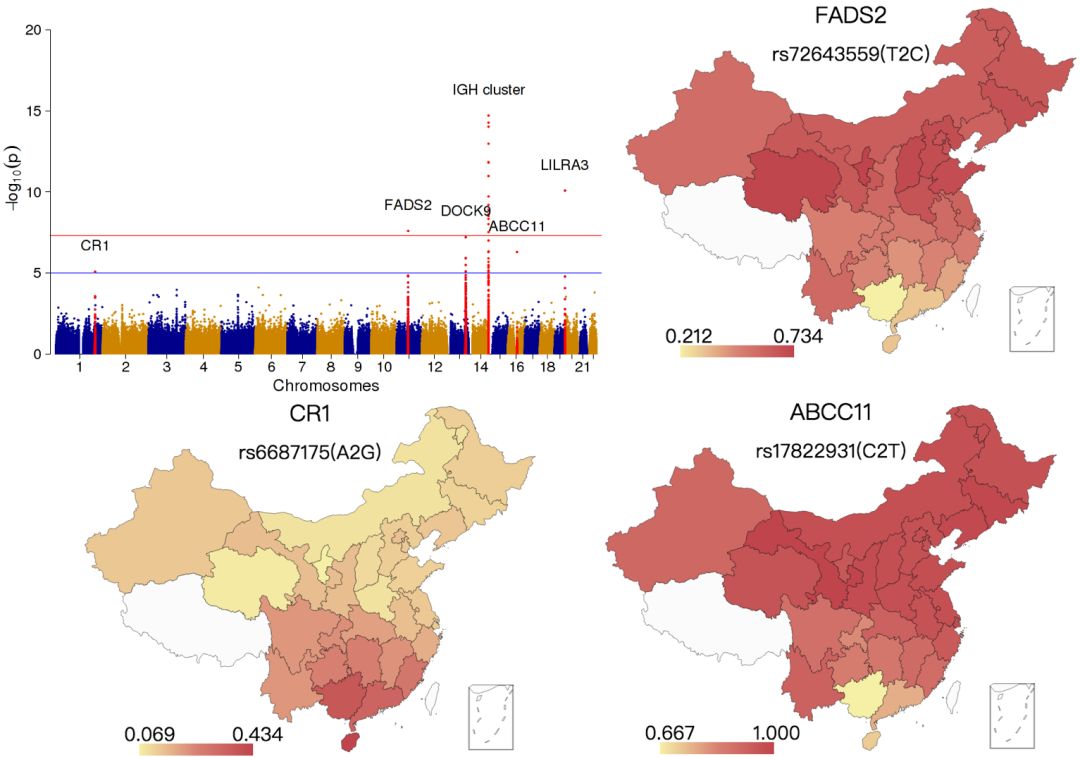 图8. 六个在中国受到显著自然选择的遗传位点以及其南北差异模式
图8. 六个在中国受到显著自然选择的遗传位点以及其南北差异模式
比如,在一个名为 FADS2 影响脂肪代谢的基因上,我们发现 rs72643559 的C型促脂肪代谢等位基因在蔬菜供应缺乏的省份(比如新疆、青海、内蒙古)中会明显富集。但和机体免疫功能相关的红细胞补体受体I的基因 CR1 却呈现了南方富集的现象。古代中国南方被称为南蛮瘴气之地,自然环境比较恶劣,病原微生物尤其是疟疾盛行,祖先们想要在这里活着,免疫力没有优势是不行的。
此外,还有一个 ABCC11 基因,这是一个与耳垢干湿、大汗腺分泌、体味(如狐臭)相关的基因,它也同样受到了选择。出过国的小伙伴可能对(那些没涂体香剂)欧美人、非洲人身上的 “浓郁” 味道多少有些印象吧?
大汗腺,又称顶浆腺,只集中分布在腋下,胯下,乳晕,外耳道里,排出的汗还含各种蛋白质和脂肪酸(听起来似乎很美味)。这些物质在体表被细菌分解之后,生成了各种不饱和脂肪酸,容易导致“狐臭”。大部分欧美和非洲人都是如此,而东亚人少有“狐臭”是因为基因突变了!
这个基因说来也有趣,本来人人都应该是大体味的(就是“狐臭”),结果却是在大约2000代之前,我们的祖先走出热带非洲,进入亚洲温带的过程中,它却发生了突变(rs17822931位点),导致大汗腺分泌减少了!带来的结果就是大部分东亚人的体味都较小,且干性耳垢。

在我们的结果中,只有广西、广东和海南中还有比较多的人保留了这个祖传的“狐臭”特征,其它省(特别是长江以北)的几乎全突变了。。。这可能是因为出汗变少,更有利于在北方温带的气候下生活——要知道祖先们可不像现在有那么多保暖的衣服可以穿——也可能这个基因在南北方被双向选择了(比如体味重,容易被猛兽发现之类的)。
其实,一直以来大家都知道中国的南、北方人有差异,但限于有限的数据,几乎没有人能够知道究竟有哪些基因带来了这些差别。而这一次对我们来说,最突出的地方在于,仅利用14万全基因组超低深度的数据就一次性清晰地找到了六个南北方有着明显选择差异的基因。这可能是第一次在中国人群体中获得了如此精细化的结果,至于当这个数据继续增加之后是否还有更多的发现,我们可以拭目以待。
复杂性状的全基因组关联研究
这次做全基因组关联研究(GWAS)的思路和策略与传统使用基因芯片数据的方法也是不一样的——更何况这还是第一次基于超低深度测序数据。
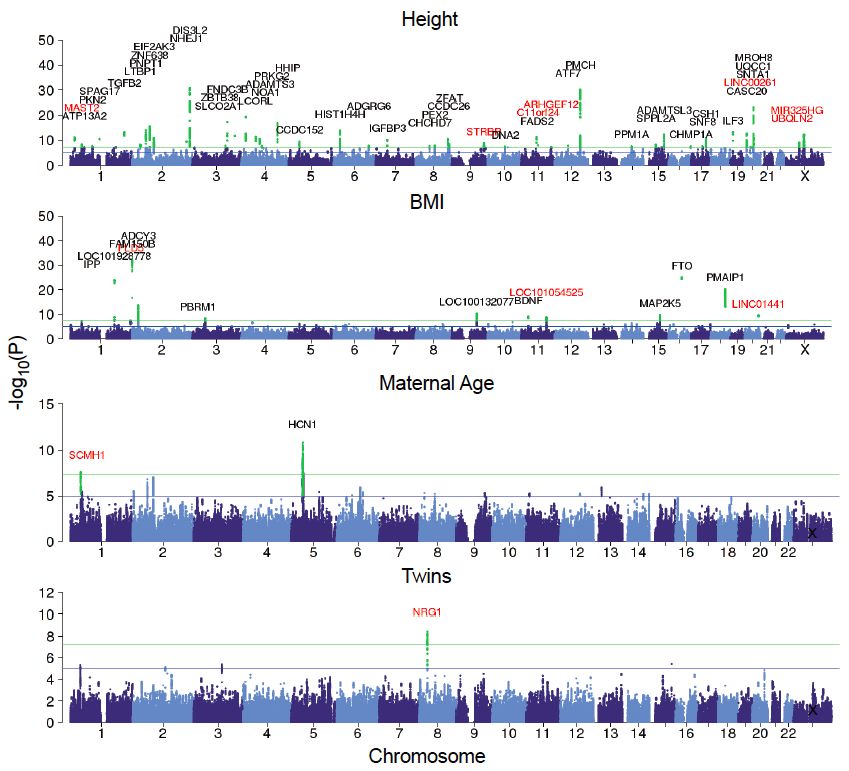 图9. 四个重要表型的GWAS研究
图9. 四个重要表型的GWAS研究
我们最终选择了4个比较有代表性的性状,其中2个是人群中的常见性状(身高和BMI)——也是已知与遗传相关度很高的性状,另外2个是与孕期相关的,因为NIPT数据本身就来自孕妇,这是很难得的一个特点。
这一块在研究开始之前,我们已经知道身高和 BMI这两个性状的遗传分析在欧洲人群中已经有比较深入的探索(比如身高的GIANT项目),但在中国人里面却还很少。
这回我们一次性发现并且验证了48个与身高以及13个与BMI显著相关的基因位点。把所用常见变异加起来,分别可以解释48%的身高遗传率以及10%的BMI遗传率。这个结果已经可以与近年来欧洲大型基因芯片人群的研究功效相媲美了。
而且,还有7个与身高以及3个与BMI相关的基因位点在世界上都属于首次发现,同时高达35个与身高以及7个与BMI相关的基因位点在中国人群中都是第一次发现。这个结果一方面填补了国家在这两项常见性状遗传研究的空白,另一方面也是又一次证实了基于NIPT超低深度全基因组测序数据在GWAS研究中的有效性。
后续甚至可以利用这些信息构建一套适合于中国人的身高预测模型,通过基因数据推断出某个人的身高情况。
而且,对于一些疾病(比如糖尿病)来说,不同BMI的病人,药物疗效会有差异,这是为什么呢?这些差异的根源是否其实就是这些有差异的基因所致?
另外两个与孕期有关的表型是:怀孕年龄和双胎怀孕。怀孕年龄是业内公认与生育力相关的一类指标,我们发现了两个与怀孕年龄显著相关的基因位点(其中一个在 EMB 基因范围内,这是一个与早期胚胎发育有关基因),这两个位点的突变与生育力密切关联。
而且,我们还在NRG1基因的内含子中发现了一个和双胞胎妊娠显著相关的突变位点(rs12056727, p=5.93E-9, odds ratio=1.99)——不能以为在内含子中的突变就不重要——它可能影响了某些基因的表达调控。在数据里这个突变的T型等位基因在怀有双胞胎的母亲中比较常见,并且会导致 NRG1 基因在甲状腺中表达上调。而且前人在用小鼠研究的时候就发现,如果敲除这个基因会导致小鼠的同胎生仔数减少,虽然暂时还没有人方面的研究,但再结合双胞胎妊娠和甲状腺机能亢进等方面的证据,其实都指向了 NRG1 的等位基因型别与双胞胎妊娠紧密关联,这很可能是影响怀上双胞胎几率的位点,当然,后续进一步的验证是非常必要的,欢迎大家一起来。
这两个方面的发现虽然已经在动物中有过研究,但在人方面却还是空白,所以这不只是填补了我们国家的空白,还同时填补了全球在这个领域的空白。
病毒谱分析
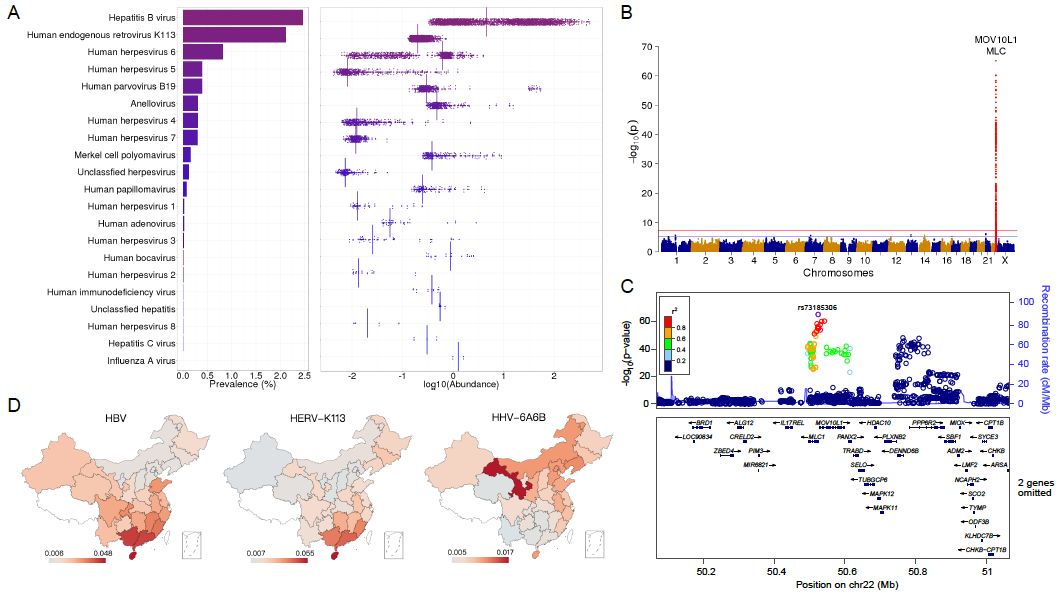 图10. 血浆病毒谱分析
图10. 血浆病毒谱分析
人体的血液中蕴含着很多有趣的信息。
最后这一部分,我们一开始的计划是研究所有的非人序列,但是发现工作量非常巨大,仅是排除环境中的微生物序列就不是一个短时间内可以完成的事情,所以我们先以病毒为突破口,研究了血浆里的病毒序列情况。这也是第一次清晰地展示了全国31个省的人群病毒感染发生率和病毒在血浆中丰度的分布(图10)。
在这个结果中,我们发现中国人血浆的病毒组与欧洲人相比差异较大。比如,欧洲人中排在第一位和第二位的分别是与皮肤急疹相关的疱疹病毒7型以及和鼻咽癌相关的疱疹病毒4型(参考J.C Venter的研究),而我们国家排在第一位的则是乙肝病毒,发生率大约为2.5%,这可能与我们国家乙肝疫苗的全面推广较晚有关。
其实这一块的结果在很多人看来到此为止也足够了。但是对于我们来说,病毒的感染情况也是一个难得的表型数据!我们最稀缺的也是这种准确的表型数据!所以,一旦算出来就不会放过!于是我们又很快用 GWAS 分析了病毒易感性和基因型之间的关系,结果又发现了另一个有趣的东西。
那就是在 MOV10L1 与 MLC 的基因区域我们找到了一个与疱疹病毒6型(HHV-6)易感性极显著相关的基因突变(rs73185306, p-value=7.3e-66, Odds ratio=3.4)。MOV10L1 编码一种结合PIWI RNA的解旋酶,我们推断如果这个基因的活性被抑制了可能会给 HHV-6 的侵入创造有利环境。但有意思的是 HHV-6 除了可引发皮肤丘疹之外,还被发现会导致中枢神经系统症状,最新的研究显示了 HHV-6 的感染与阿兹海默症(老年痴呆症)显著相关。
而 MOV10L1 的突变则是我们第一次发现的,之前发现HHV-6与中枢神经系统症状有关的研究并未发现这个突变。所以我们觉得或许存在另一个可能,那就是 HHV-6 其实并不是直接导致中枢神经系统症状的原因,而是这个基因的突变所致,只是恰好这个突变发生了之后,这些人也变得更容易感染 HHV-6 而已,所以就误以为 HHV-6 和中枢神经系统症状相关(这是本文的假说,我们文章没写明这个猜测)?如果这个研究能够进一步得到证实,那么就有可能通过检测一个人是否携带这个突变(可能同时表现为易感HHV-6),推断其罹患阿兹海默的几率。
那么,这些都有什么意义?
我想,能够看到这里的小伙伴应该不难想象这些研究的意义。总的来说有5个:
第一,我们构建了一套新的研究思路和方法论。解决了当前组学领域难以利用超低深度全基因组测序数据进行一系列遗传学研究的局限和挑战,并成为领域中第一个证明了NIPT数据能够用于回答诸多遗传学以及医学领域重要问题的成果(只要足够准确的表型数据)。这些方法和策略在遗传学以及医学基因组学中都有被参考和应用的价值;
第二,我们提供了一种新的组学研究思路,让大家意识到低深度测序数据对于大规模的组学基线研究来说是可行的。并且第一次证明了超低深度(<1x)全基因组测序数据同样能够开展许多高质量高水平的组学研究;当然,如果能够测到1x-2x(但不一定需要更多了),那么结果相信会更好;
第三,这个课题所获得的一系列成果包括群体变异数据、群体遗传学、GWAS和病毒谱,以及已被验证和尚未被验证的结果,填补了不少国内外的研究空白,相信对后续的研究也会有启示作用。同时这个用于研究的14万数据本身也是一个良好的普通中国人基线代表。这些成果将可以在肿瘤、复杂疾病、罕见病等的研究和解读中充当背景代表;
第四,数据的自主权利,见下文;
第五,这是组学研究从单体检测到大规模人群研究模式的跨越,这是一个新的趋势。
这个课题大约历时一年半(加上伦理审查、立项等的时间则是2年),经过各种努力,如今得到了一个还算不错的结果。另一件值得一提的事情是,在文章的投稿过程中,为了更好地遵守《人类遗传资源管理暂行办法》,就跟《Cell》沟通说,我们的数据按照人遗规定都不能外传,只能留在深圳国家基因库。当然这个过程并不容易——这是打破原有规则的做法——文章也完全有因此被拒接的风险,但最后《Cell》还是同意了。所以,我们所有的基因数据都不需要往NCBI、EMBL、DDBJ等海外数据库备份,这个应该是第一次发表组学文章可以这样做,这无疑也是一个非常具有战略意义的先例。
这几天有些网友看到我们竟然可以不需要传数据也能发《Cell》,有些难以相信,甚至还有错误报道的阴谋论者。我作为第一作者想说的是,不用怀疑,我们就是做到了,而且开了先河打破固有规则,你应该高兴才是,中国很强,我们团队也不弱。这个数据不但没有传输至海外数据库,就连通讯作者中两位给我们提供很好建议的国外教授也没能见到数据,只能看到我们做出来的图表结果。
晒一下课题组研究团队核心成员的合影:

研究团队核心成员合影 左起:刘斯洋博士(第一作者),徐讯博士(通讯作者),陈芳(共同第一作者),金鑫博士(共同通讯作者), 黄树嘉博士(共同第一作者)
再来一张课题组主要生信团队的合影:

课题组生物信息团队主要成员合影 左起:刘荣,林珑,刘斯洋博士 (第一作者),李子龙,黄树嘉博士 (并列第一作者)
做完这个项目之后,其实我们自己也发掘到了更多有意思的问题,只是限于数据的种类,我们自己也暂时没办法很快对其进行验证,这是接下来要解决的地方。
P.S.
这个项目最终能够成功,除了我们团队的努力之外,也少不了华大内部各个部门同事之间的高效协作以及领导们的支持。另外,在今年4月份,我们其实就已经在Cell的服务器上预发布了这个成果,不知道大家是否有看过:迄今最大规模的中国人基因大数据研究成果在Cell上预印。那时因为一切还在未定之中,我甚至都不敢转发,有网友也表示不看好这个成果。
这个文章从今年3月份投稿之后就正式被 Cell 送审,中间也只按Reviewer的意见小修了一次,到7月16日就接收了,速度应该不慢,但直到现在才正式发表,除了排刊,可能也与 Cell 官方专门做了采访有一定关系吧。
更多
- Siyang Liu, Shujia Huang et al., Genomic Analyses from Non-invasive Prenatal Testing Reveal Genetic Association, Patterns of Viral Infections, and Chinese Population History, Cell (2018), https:/doi.org/10.1016/j.cell.2018.08.016
- Cell官方报道:https://www.eurekalert.org/pub_releases/2018-10/cp-sup092718.php
- 纽约时报报道:https://www.nytimes.com/2018/10/05/science/china-genetics.html
- China.org报道:http://www.china.org.cn/china/2018-10/05/content_64891101.htm
- 华大公众号报道:https://mp.weixin.qq.com/s/xmiOFfax77W9pJ5kUmtSwg
- 央视报道:https://mp.weixin.qq.com/s/Z5-9Jddq2e3a7n-BsQEVQA
- 国内数十家媒体报道:https://mp.weixin.qq.com/s/cUMIbdSwns23GdQRni9pSA
- 大规模中国人基因频率数据库(CMDB):https://db.cngb.org/cmdb/
- 收录于“人在知乎,刚发《Nature》”
订阅
关注我的个人公众号:helixminer(碱基矿工)
