
2024 年 1 月 31 日,农历冬天的最后一个月,我们广州出生队列基因组学研究的文章——“The Born in Guangzhou Cohort Study enables generational genetic discoveries”[1]——正式在 Nature 主刊上发表了!这是广州出生队列第一个里程碑意义的成果,也是我第一篇作为第一作者发表到 Nature 主刊上的文章。文章上线时 Nature 做了两个篇独立的报道[2,3],对我们的成果做了评论,感兴趣的朋友可以查看文末的『原文链接』。

Nature 文章截图(第一作者:黄树嘉,刘斯洋,黄铭曦和何健荣;通讯作者:邱琇和夏慧敏)
这里我不打算重新对新闻报道的内容做赘述,我们队列的公众号也有报道。这里,我想做更专业、深入和完整的解读,讲讲这项研究的具体发现,以及项目过程中我的一些思考和体会。希望这可以更符合关注了我这个公众号的朋友们的口味,文章有些长,本想拆开成两篇的,想想还是算了,就做一篇吧,这样才连贯。
我喜欢从事生物信息学和生命科学研究,它既不枯燥又充满挑战。这个项目的进展并非一帆风顺,整个过程解决了很多问题,从我们讨论确定大致的研究路线到实际执行和最终发表成果,用了将近三年时间,其中审稿就花了一年半,来来回回审了 5 次(包括编辑的最后一次校审)!这是我截至目前为止历经过最漫长的审稿时间,中间我不止一次担心是不是要杯具了。
从整个文章的结构来说,归结起来,我们主要做了如下 4 件事:
- 完整构建了广州出生队列(BIGCS)自然人群的基础遗传图谱;
- 从群体遗传学的角度,把南方中国人(主要是粤语人群)的遗传历史讲得更清楚了;
- 母婴性状 GWAS 研究以及年龄特异性遗传效应的发现;
- 实现母婴遗传效应分离的方法并建立跨代孟德尔随机化分析方法。
研究背景
为什么要做这项研究?主要是两点:
- 疾病是环境和基因互作的一个结局。基于大规模前瞻性的出生队列基因组研究将是一个好的策略,它是理解基因和环境如何互作从而影响人类健康的重要研究手段,但这样的研究在中国乃至整个亚洲都是稀缺的。
- 胎儿生长发育受到母体宫内环境和胎儿自身遗传的影响,但是,宫内环境和遗传效应对胎儿的生长发育到底起怎样的作用尚不清楚,另外也缺乏有效揭示宫内环境对胎儿带来影响的方法或者手段。
广州出生队列
广州出生队列(Born in Guangzhou Cohort Study, 简称 BIGCS)从 2012 年启动至今已经整整做了 12 年——应该是我们国内较早的一批自然人群出生队列,取得了很多好成绩,这些已不需我来重述。但我们一直希望能够在基因组学研究上有所突破,流行病学研究并非总能有效回答在疾病和性状上所发生的现象,我们希望可以从更加根本的遗传特征上去寻找关键信息。但这得一步一步来,BIGCS 第一步是先从入组人群中随机抽取了 2000 个健康的母婴对(其中原计划是包含 400 个家系),总共是 4,215 个人(我必须说这个样本并不算大),考虑到测序成本的原因,只做了 7x 的全基因组测序(WGS)。2020 年 6 月我加入了广妇儿,来到了出生队列研究室的团队里,并从那个时候开始正式投入到这个项目中。在这个过程中,我首先从零开始搭建了整个基因组学分析框架和流程以及项目过程中的分析方案(与技术细节),与刘斯洋教授制定了初步的研究路线并与邱琇老师以及团队里的其他人共同讨论,最终确定了整个项目的故事线和方向。
这个项目采用的是大样本+低测序深度的策略,对自然人群的组学研究来说,我会认为样本量对于科学发现而言是更加重要且更具有决定性的因素。所以在同样的件下,应该检测尽可能多的样本,虽然如此我们的第一阶段也只能做到 4,215 例。再加上每个样本只测了 7x——这个深度应该是参考了千人基因组计划的策略——最终原始数据在经过我质控之后平均深度就只剩下 6.63x 了,我同时做了一些数学上的推导分析了这个深度的合理性以及它的潜在挑战,如下图:
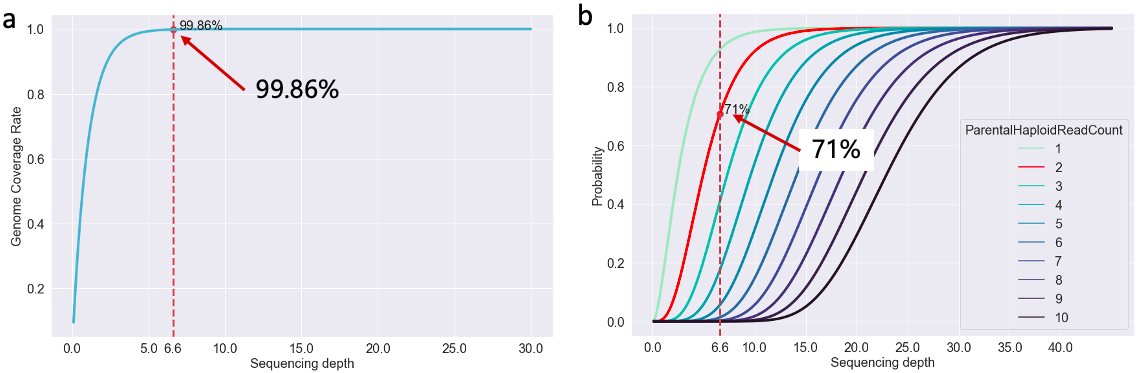
图 1. 测序深度与基因组覆盖率
这里的 图1.a 是不同测序深度条件下,对基因组的覆盖率,可以看到 6.6x 的深度理论上其实已经能够覆盖整个基因组 99.86% 的区域了,10x 以上时更是无限接近 100%,所以从数据的覆盖度来说这个深度是 OK 的。但光看这个是不够的,因为我们是要进行变异检测的,最终分析的落脚点是在突变数据集上。
对于突变数据来说,我们要准确全面地检测出来,就得确保在两个同源染色体(人是二倍体,有来自父母的两个同源染色体)上能测到足够的 DNA 序列,否则就可能漏掉信息导致变异检测不准确——特别是对于杂合突变来说更是如此。所以,我分析了在不同的测序深度条件下,基因组上任意一个位点都能被测自两个同源染色体的 read 覆盖 N 次的概率(图1.b)。我觉得每一个位点起码要被两个同源染色体覆盖分别两次才可以在变异检测的时候得到较准确的结果。在图里我们可以看到,对于 6.6x 的深度来说,理论上只有 71% 的位点符合这个要求(红色曲线),应该说对于基因组的纯合突变来说问题不大,但对于杂合突变的检测而言,就可能有 ~30% 的杂合突变位点会出现 杂合丢失 的假阴/假阳性结果(当然我的假设条件较严苛,实际比例应该不至于如此高),这个也是我后面必须想办法解决的地方。
当然理论分析也仅仅只是数学计算,最终情况还是要看真实数据的表现的。但理论分析的好处就是,可以让我们心里有一个衡量标准,而不是两眼一抹黑,然后瞎搞。
我们都做了哪些研究
那么,接下来我将逐一解释我们主要都做了哪些事情,以及为什么要这样去做。
1. 构造分析流程,完成变异检测,构建 BIGCS 自然人群的基础遗传图谱
第一步自然是要完成所有样本的全基因组变异检测,获得准确的突变数据集,这是实现所有下游分析的关键。
刚开始时我们没有任何关于基因组数据分析流程,团队除了我之外其他人也没有足够的经验。如果只是为了完成一两个人的全基因组数据分析,其实也不麻烦,参考 GATK 的文档也能做下来,但我们是四千多个样本,中间分析步骤繁多,产生的数据文件更是成千上万,手动检查是不可能的,所以除了核心步骤之外,要完成这个事情就还得具备对任务完成情况进行监测的能力。为此,我从头设计并实现了一个适合于大规模 WGS 数据分析的半自动化流程(如下图 2)——ilus,我在公众号里也分享过:ilus:一个轻量级全基因组(WGS)和全外显子(WES)最佳实践分析流程生成器。有了这个之后,即便只是一个人也可以轻松搞定这个分析。
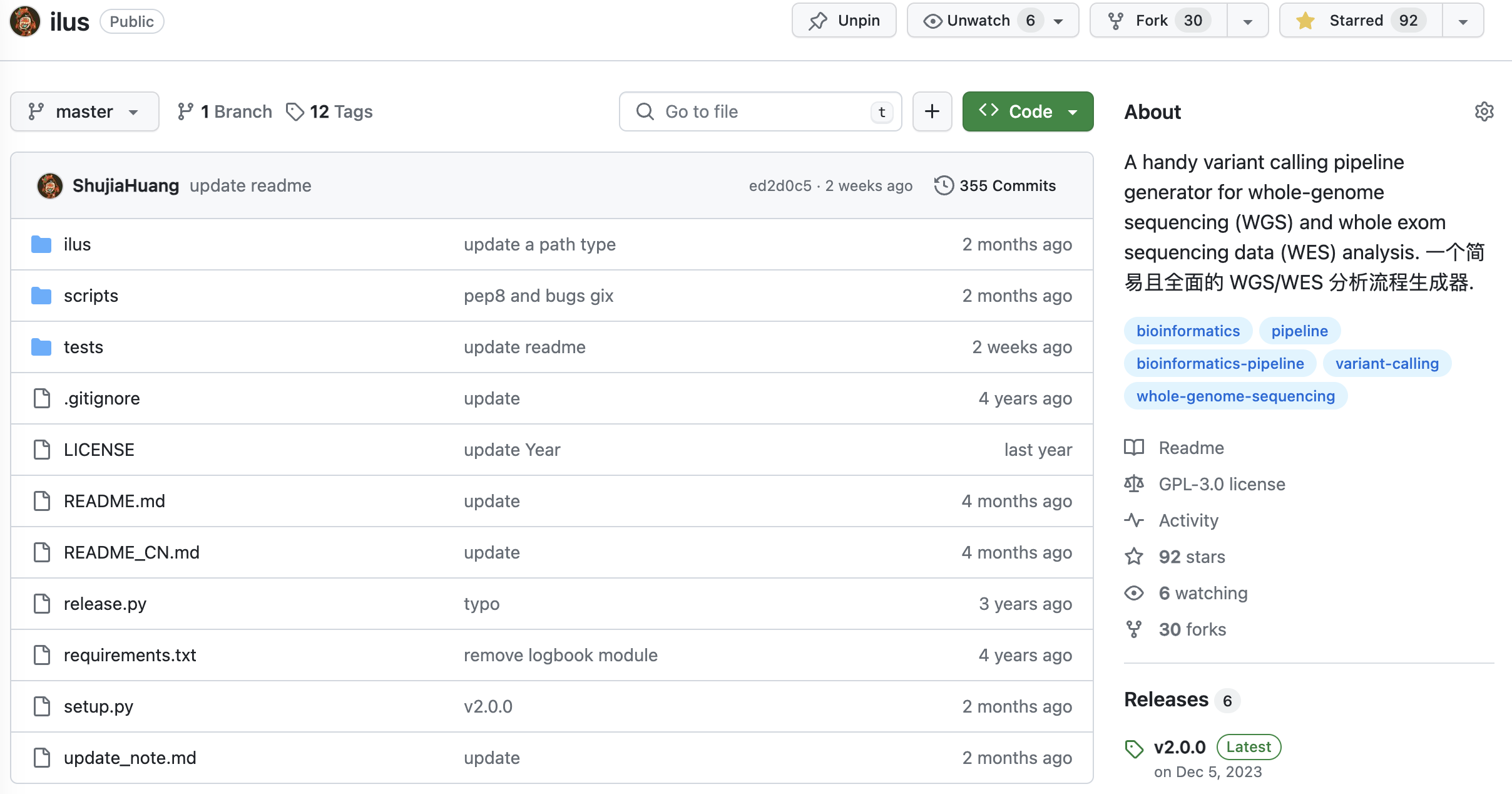
图 2. WGS 分析流程(https://github.com/ShujiaHuang/ilus)
目前 ilus 已经到了版本 2.0,除了 GATK 最佳实践之外还兼容了 Sentieon,并且可以进行 WES 或者其他捕获型测序策略的数据分析流程的构建。如果可能的话,我特别推荐你用 Sentieon。
过程虽然繁琐但就都比较常规,我就不再赘述,Nature 文章的方法部分对此也做了很详细的描述。但在对突变数据集进行深入质控方面,我想特别指出的是一定不要只做一个 VQSR 就完事了——我的这个判断应该可以适合大多数 WGS 项目,对于低深度 WGS 更是如此。仅仅只是 VQSR 得到的结果和芯片数据比起来质量是不行的——我们的数据显示 SNP 还不足 95% 的一致性。
原因其实我也知道,主要还是来源于上面理论分析时提到的杂合丢失,如何解决这个问题呢?以前在用 NIPT 超低深度数据进行 14 万人的遗传学研究时,我就已经知道,这个时候进行 Imputation 将是一个合理的选择。但是,这里不应借助 reference panel,否则数据会被局限在 panel 之下,这对我们是不但得不偿失,而且不正确。所以,我自然地想到了通过 reference panel free 的方式来进行——也就是不用 panel 了,仅仅依靠自己数据的情况来进行推断。但要考虑到深度低,每个样本此刻的基因型会具有一定程度的不确定性,因此在重新填补和推断的时候应该用基因型似然概率(genotype likelihood)而不是直接用 genotype,这也就是我最终的解决路线,这个步骤我们称之为 LD-Based refinement,因为这其实是通过连锁不平衡来实现内部基因型填补和校正的。那么最后效果如何呢?可以说非常不错,如下图 3:
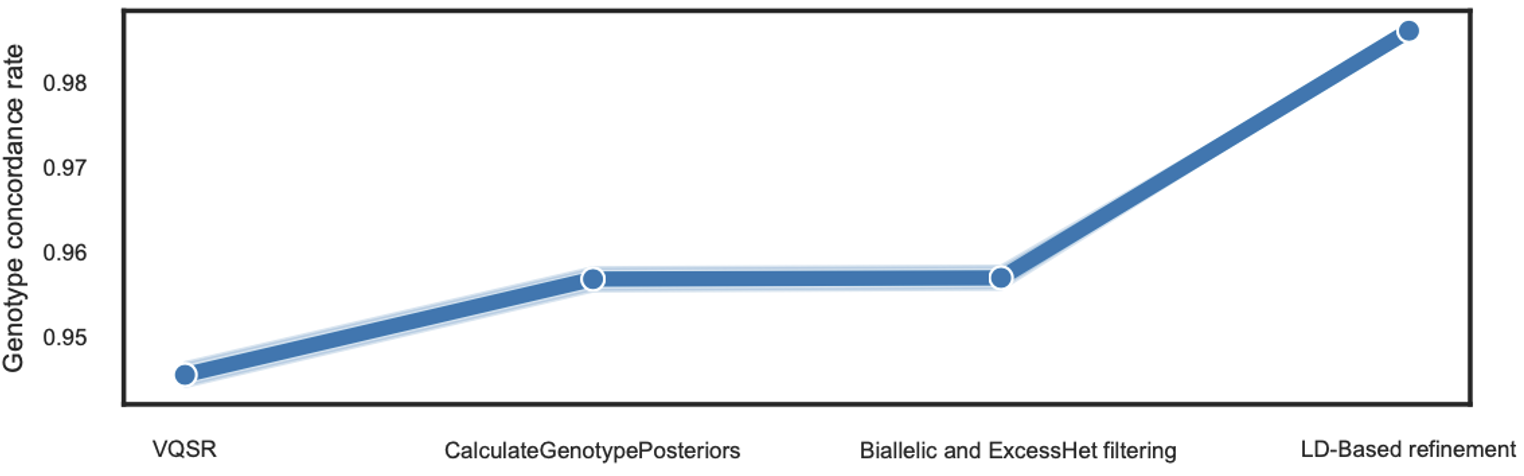
图 3. 变异质控
可以看到 refinement 之后,基因型的整体一致性就达到了 99% 以上,这就符合预期了。当然,这个过程中还有其他细节,文章里也有详细说明,但比较细枝末节,我就不在这里重述。
最后,我再提一嘴,我们原本的样本一共是 4,215 例,做完初步变异检测之后(还未质控),我们对样本的测序质量、性别一致性、亲缘关系一致性等等各个方面都做了层层过滤,最终就只剩下了正文里说到的 4,053 例。
其实,我上面定下的突变数据过滤条件十分严格,被过滤掉的数据很多,大约 33% 的原始突变数据被过掉了(最后剩下 56M 个群体突变位点)——是有些可惜的,但我也不想得到假的东西。有了这个干净的突变数据集之后,我们还用它构造了一个新的特别适合于中国人群的参考图谱(reference panel)可以为研究人员提供更好的基因填补(Imputation),并将它做成了在线的数据库,名字是:Genome Database of Born In Guangzhou Cohort study (简称:GDBIG,http://gdbig.bigcs.com.cn/),对于有需要的朋友直接到这个网站注册就可以使用了。这里我吸收了之前我在华大构建 Chinese Millionome Database(CMDB)的不足,把这个 GDBIG 做得更好了,你如果用了应该可以体会到,功能也更多(包括,突变频率查询、基因型填补和特定母婴表型的 GWAS meta-analysis 等),还可以通过 API 在命令行下查询。
GDBIG 主页
2. 群体遗传学分析进一步揭开南方中国人的遗传历史
对于广州出生队列的亲子对基因组数据,原本我们并不需要将群体遗传学作为重点内容来凸显我们的特色。我们之所以还是要这么做有两个原因:
第一,项目刚开始时,我也想将焦点集中在 GWAS 上,但我做了功效分析(Power analysis)之后,很快就意识到风险太大了,样本量不足,恐怕只能发现一些别人发现过的信息(而且还不一定可以看全)。初看我们有四千多个样本,似乎不少,但实际上如果要做 GWAS,母子需要分组,这样一来,母亲组和小孩组分别就都不到两千人了,结果会如何我没有信心。所以,最好是能有其他的突破口,这个时候人群的群体遗传研究就成了一个重要的选择,而且这恰好是南方中国人群,此前研究并没有很好覆盖到,那么做得好的话是有可能有新发现的,填补一些空白也将是有意义的。
第二,作为潮汕人,我其实非常好奇南方中国人的群体遗传特征,特别是广东省三个主要方言人群之间是否有什么有趣的特点。我们的随访数据也有很好的方言信息,刚好适合做这个事情。
所以,群体遗传学分析自然就成为我们重点要探索的一个方面。
我们也的确有了一些新的发现,首先是在 PCA 分析中,我们发现遗传信息与方言的匹配度要显著高于籍贯和民族,这其实并不让人感到意外,也许你不做这个分析也会觉得应该如此,但我们用数据给出了直接的证明。而且,这应该是第一回用人群全基因组数据把中国人中 10 种主要的方言与他们的遗传信息建立了联系(图 4)。眼尖的朋友应该还隐隐可以看出,这个图谱和方言人群在地理上的分布十分相近。我想这对于中国传统方言的研究将是有积极意义的。
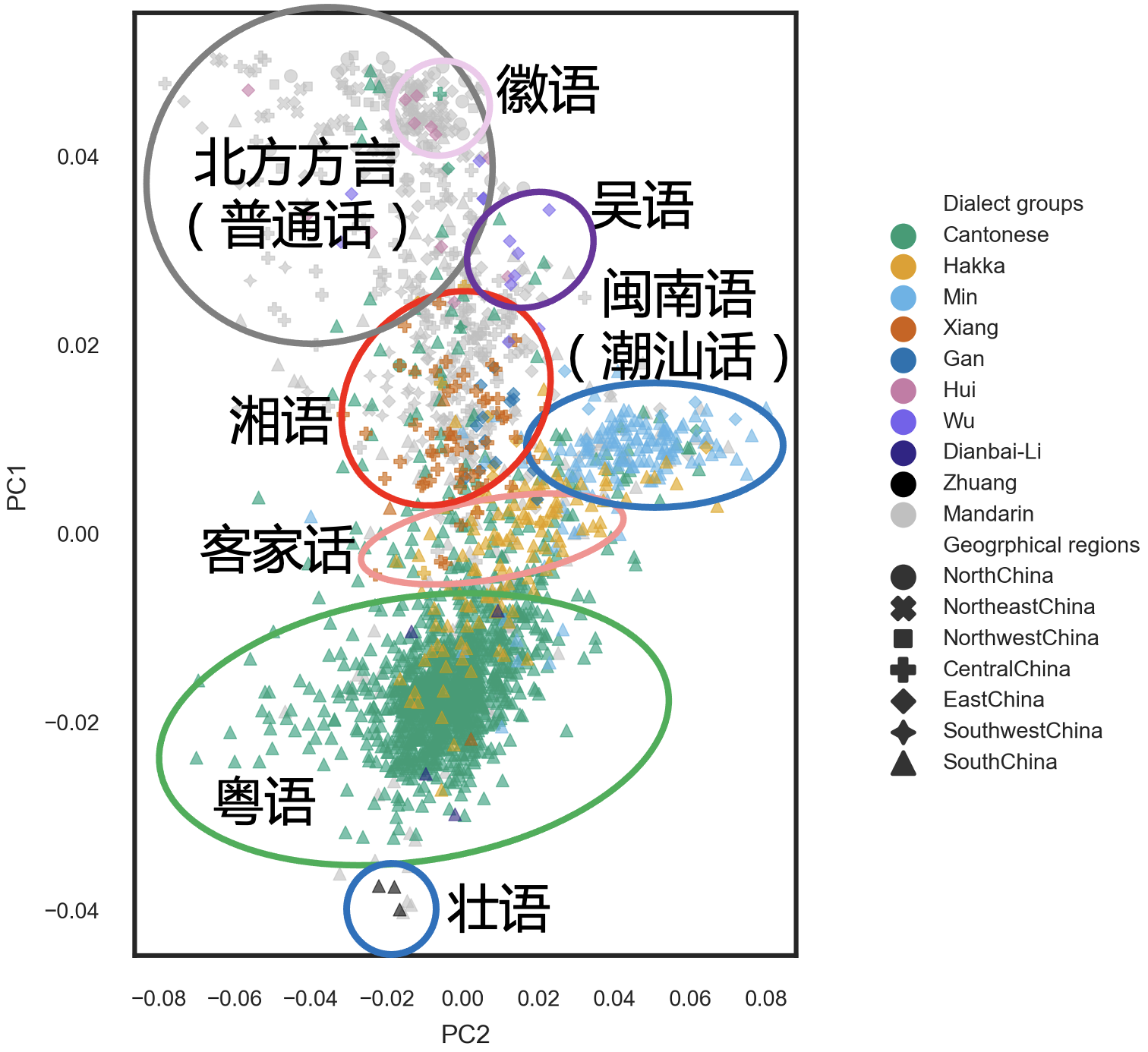
图 4. PCA
有了上面的分析之后,我们就想往历史深处看得更远一些。我们十分好奇,是否还可以从遗传学角度推断出更多和这些方言有关的人口统计学历史(demographic history)?看看我们的方言祖先们在历史的长河中是否发生过什么有趣的故事。那么,说干就干,考虑到样本中方言人群的样本量问题,我们就以五个样本量大于 50 的方言群体(普通话、湘语、粤语、客家话和闽南语)作为代表开展研究。
经过一番分析之后,我们清楚地看到这五个方言群体之间的人口动态变化是相似的,都大约在 15 万年前进入人口瓶颈期,有效人口数不断减少,并大约在 2.5 万年前达到最低点,随后才开始逐步恢复(图 5a)。为什么会这样?我查找了该时段的地球地质历史,发现该时期恰好是地球的末次冰期,并且在大约 2.65 万至 2 万年前达到冰期顶点——即末次盛冰期。在这一时期中,地球上的冰川规模达到最大,极端严寒和干燥的气候导致生存环境变得极其恶劣,有效人口的大幅减少就发生在这一时期(从全球其他人群的基因组上同样可以观察到类似情形),之后才开始逐渐恢复。
另外,我们还计算了这五种方言群体之间的遗传分化时间,发现他/她们的分化始于新石器时期(史前一万年),并在大约 3千-4 千年前逐渐稳定下来,而这个时间恰好与中国古代农业社会体系的形成和发展相吻合(图 5b)。
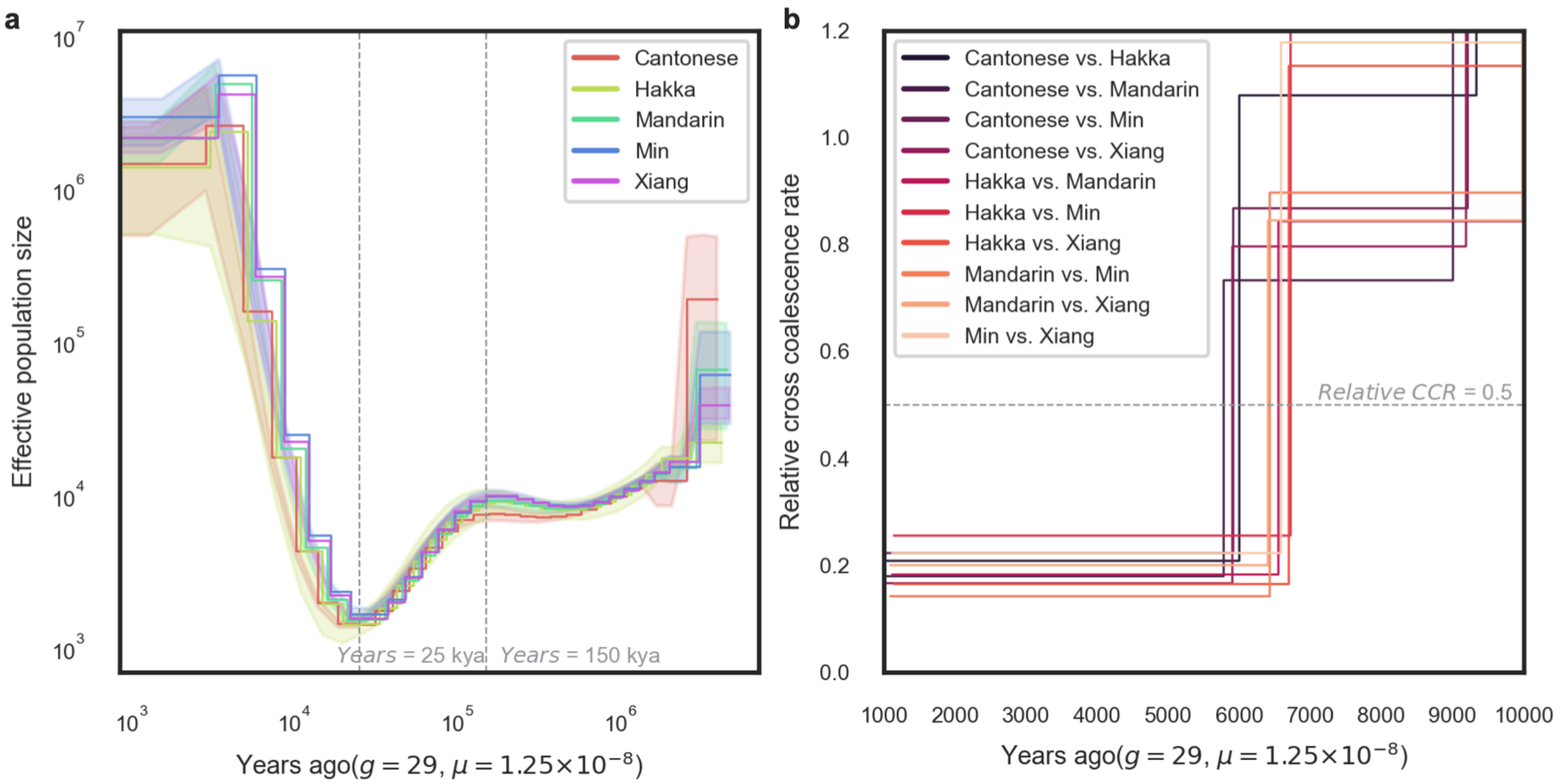
图 5. 通过 BIGCS 基因组数据推断中国人的人口统计学历史
最后,既然粤语占优,我们当然希望再看看这个群体的其他特征。首先是结合古人 DNA 做了 ADMIXTURE 分析,我们可以非常直观地看到,生活于南方的粤语人群主要是中国北方人(包括黄河流域一带的北方古人,图中橙金色)与南方壮侗语系人群(Tai-Kadai,图中浅蓝色)的混合体,并且有更明显的北方古人痕迹,而南方古人的成分则很低(图中深蓝色)——也就是说粤语人的祖先应该是北方古人(图 6)。当然做出这个判断并非只靠 ADMIXTURE 分析,而是还进行了其他的分析,特别是 f4 statistics 结果的支持(这里要特别感谢付巧妹老师团队在这一块的分析和解读)。
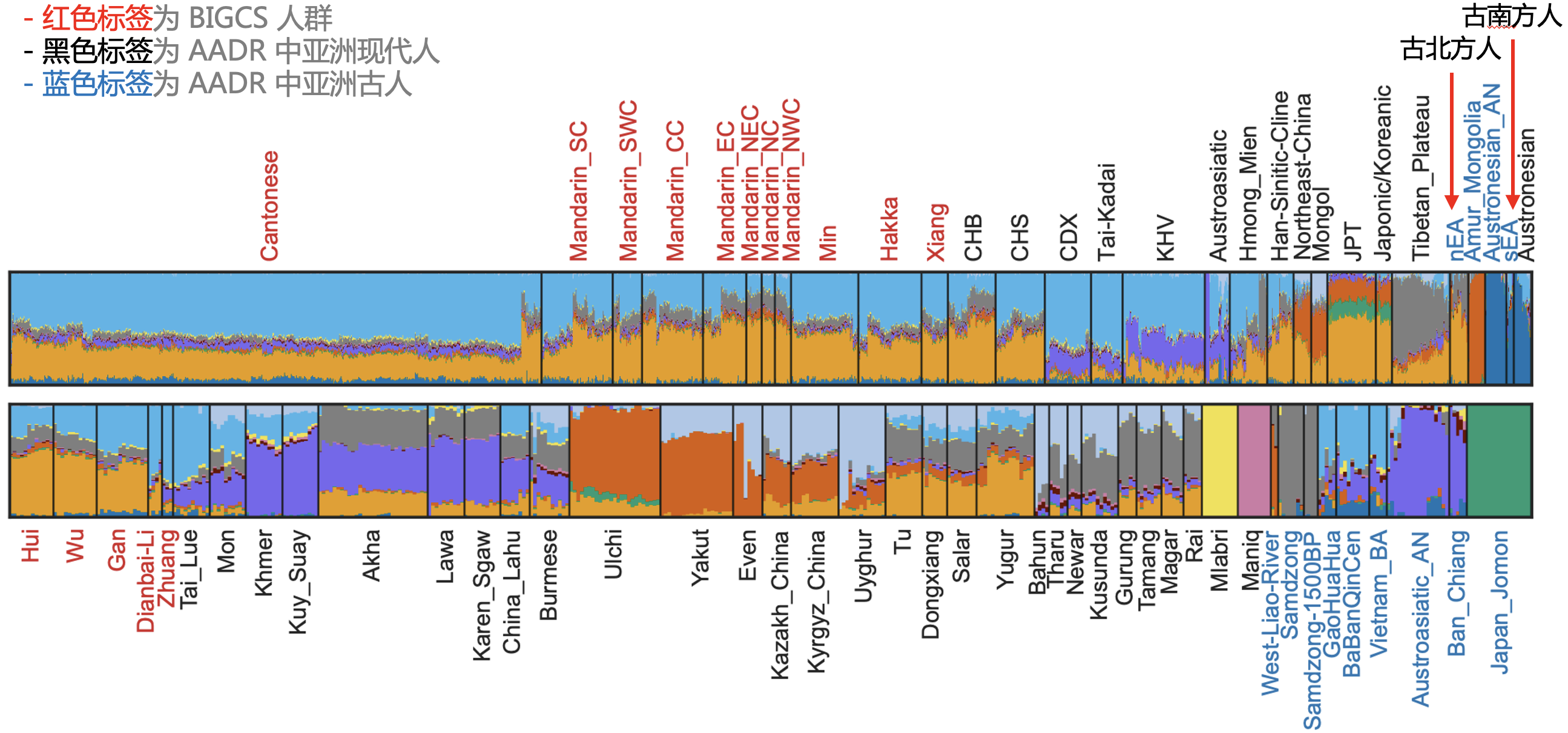
图 6. ADMIXTURE 分析
既然我们看到粤语群体的祖先大概率来自北方,那么一个顺理成章的问题就是,他们是什么时候南下的?我们通过计算人群开始发生遗传混合的时间点来推断南下时间(图 7)。
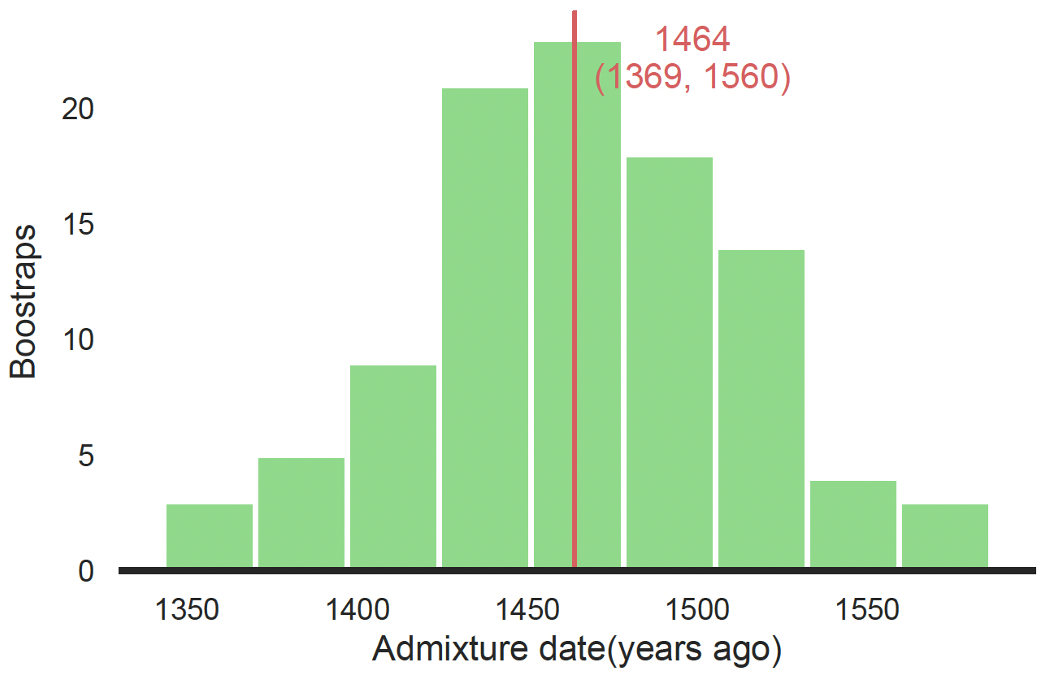
图 7. 粤语人群南迁时间推断
结果我们发现,这个时间大约是 1,464 年前。但要注意这个时间要从出生时算起,按照 BIGCS 的年龄结构,要往前推大约 30 年,考虑到样本入组和测序时间,我们从 1990 年算起应该比较合适。所以,粤语人群和南方人发生混合的时间大概是公元 1990 - 1464 = 526 年(南北朝时期)。但是,这已经是和当地人发生基因混合的时间点,人群迁入时间则还要再往前推一些,可能是始于五胡乱华、中原汉人被迫“永嘉南渡”之时,这是一场前无古人的大迁徙,这个时期西晋朝廷渡过长江,建立东晋政权,此时同宗同族的人们抱团取暖,社会强调血统与身份,家族传承形成书面谱系。东晋朝廷依据父系家谱,判定官员的品阶甚至发展出“谱学”——“上品无寒门,下品无势族”(出自《晋书·卷四十五·刘毅传》)。
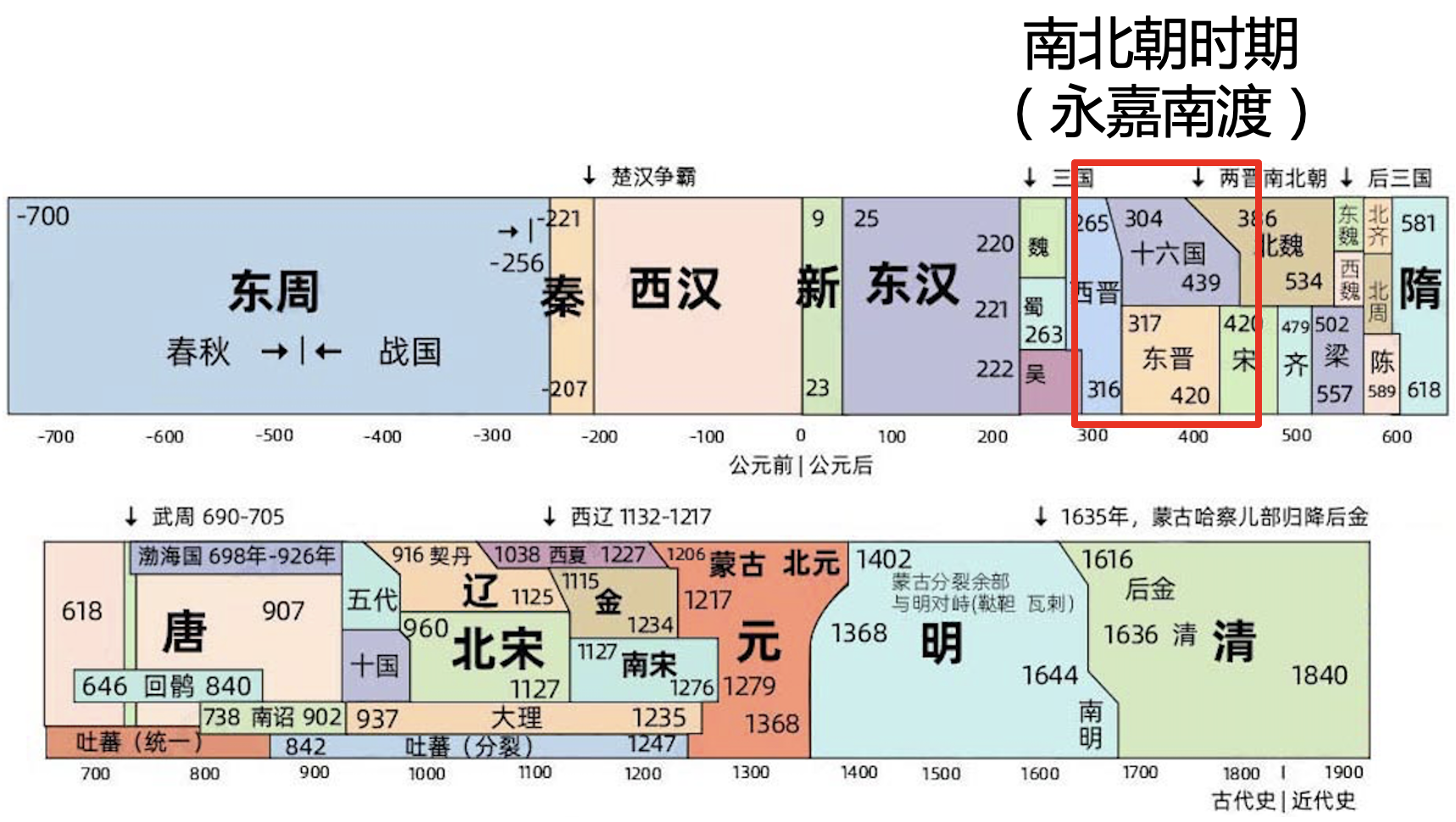
图 8. 中国历史朝代演变图,图源:亿图图示
“六朝烟水间,金陵崛起”,从这一刻起,中华大地成了民族与文化相互融合的大熔炉,中华文化所占有的核心空间得到扩张!而曾经的历史事件或多或少地镌刻在我们的 DNA 之中。看着基因组上呈现的信息,为了找到更多可以印证的历史事件,我也翻阅了一些历史文章,看着魏晋南北朝各个朝代变更的阴谋与屠杀:轻则灭门,重则亡族;因皇帝一时的喜好便生灵涂炭,因一刻的厌恶便诛灭功臣。整整三个世纪的纷乱,北方的百姓有时候好不容易逃难到了南方,即使同是汉人,也会被南方的士兵因为政治和军事管理问题而全部杀尽。死去的人数越多,就越对数字麻木。
三个世纪的纷争,现在说起来就像是没有感情的事件,但对当时的人们来说,桩桩件件都是生离死别,影响一生命途的变故。
青山依旧在,几度夕阳红?
从宏观来看,我们就是时间中的沙尘,一辈子的悲欢喜乐,不过是漫长时光中的一瞬。作为草芥的我们,是人类血脉与文明的延续。活着,传承血脉,就是生命最大的意义和价值。
说得远了。
回观基因组上的信息,这波澜壮阔的历史,我们无法详细描述——需要更多考古和组学证据的关联,虽千言万语也只能浓缩成文章补充材料(Supplementary)里的一句话:the admixture event occurred approximately 1,464 years ago (95% CI: 1369-1560 years ago), coinciding with the period of the Northern and Southern dynasties in China (Fig. S11, Table S7), which was a particular period of turmoil and warfare in Chinese history(这不免略有些遗憾,但我们目前确实做不到历史的展开,也期待之后可以有更多的研究)。
总的来说,通过这些结果,我们把南方中国人的遗传历史讲得更加清楚了,也为理解华南地区方言和人口演变的相互关系提供了积极的线索。
3. 多个母婴表型的全基因组关联研究(GWAS)与年龄特异性遗传效应的发现
虽然样本量不多,但 GWAS 依然是我们这个研究中的重点。我们挑选了 12 种妊娠期表型和 6 种婴儿表型进行了分析——分为成人组和婴儿组各自进行 GWAS,除了重现了部分已知的结果之外,我们还很幸运地有了两个方面的新发现!
第一,我们发现了 3 个新的遗传关联位点,其中两个与母亲特征(总胆汁酸-TBA和妊娠期体重增加-GWG)相关,一个则与婴儿脐血低密度脂蛋白(LDL)水平有关(图 9)。
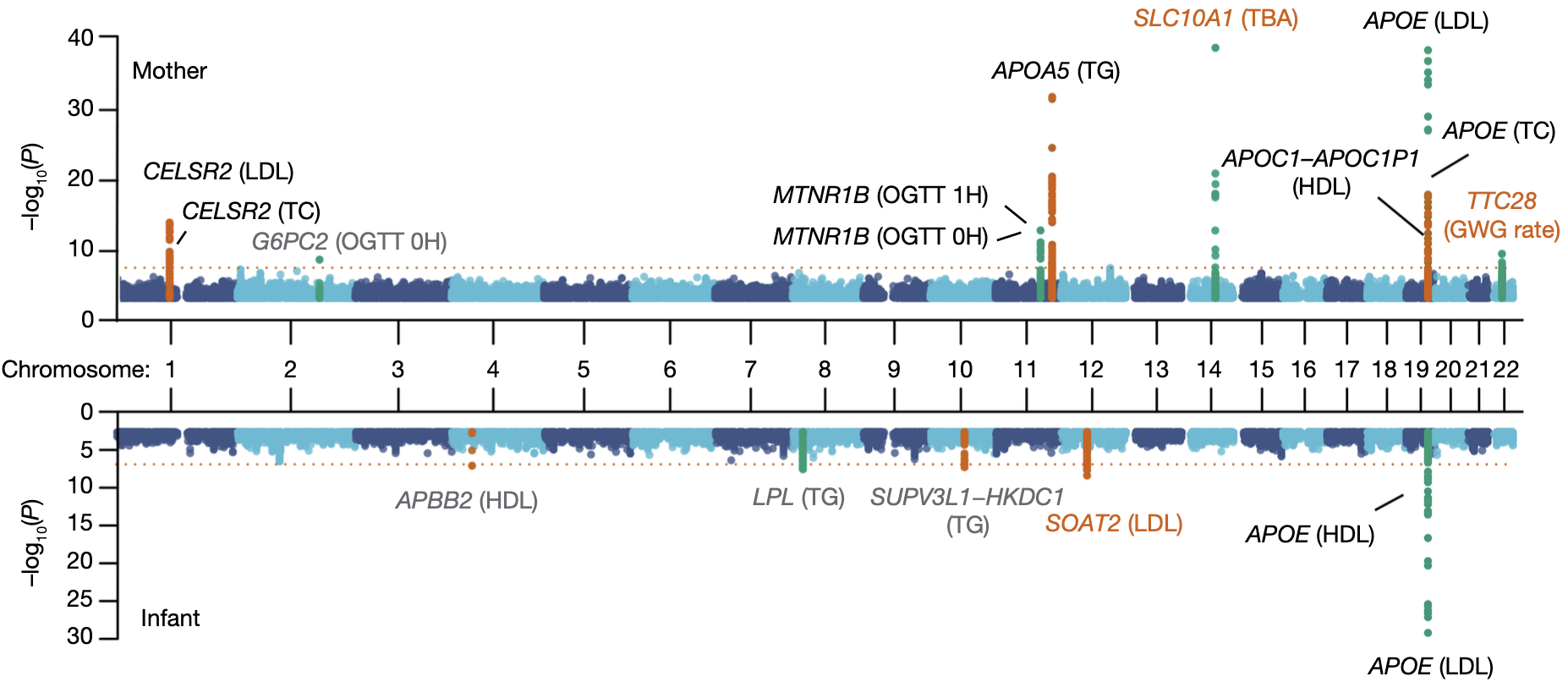
图 9. 母婴表型的 GWAS 分析结果(图中朱红色字体标注的是我们新发现的关联位点)
这其中最有趣的是和胆汁酸相关联的位点——rs2296651,它发生在 SLC10A1 基因的编码区,是一个错义突变,会导致编码的丝氨酸(Ser)转变为苯丙氨酸(Phe)(p.Ser267Phe),从而引起功能异常。我们调查发现,SLC10A1 基因编码的蛋白属于钠/胆汁酸协同转运蛋白家族,它是参与胆汁酸肠肝循环的完整膜糖蛋白,主要负责将被动吸收初级和次级胆汁酸运回肝脏。我们进一步分析发现,携带 rs2296651 突变会使妊娠期间胆汁淤积的风险增加 3.55 倍(OR 值是 4.55)。这个基因位点的突变除了会引起或加重孕妇胆汁酸淤积症之外,还可能引起(或加重)新生儿原发黄疸的程度。
人体中排入肠道的各种胆汁酸 95% 以上是要被重吸收的。重吸收的胆汁酸经门静脉入肝,肝细胞再重新摄取,转化为结合型胆汁酸再次排入肠道,形成胆汁酸的肠肝循环(enterohepatic circulation)(图10),每天6-10次[4]。
图 10. 胆汁酸肠肝循环示意图。图源: https://www.mdpi.com/1422-0067/22/3/1397
刚开始的时候,我以为这不过是寻常的新关联位点而已。但当去看这个位点在全球的突变频率分布情况时,我注意到它竟然只在东亚(特别是东南亚)地区出现,而且呈现明显的南高北低的分布现象(南方突变频率达到 10%,而北方只有~2%)(图 11),这很可能意味背后存在某个/些因素导致它出现正向选择的倾向,否则一个突变位点几乎很难呈现这样的分布规律。所以我做了更多的调查,发现这个基因编码的蛋白原来还是乙肝病毒(HBV)感染人体的一个受体[5,6]!想来其实也合理,这个基因编码的蛋白将胆汁酸运回肝脏,而 HBV 偷偷搭了便车一同混了进去。凑巧的是这个位点的突变,恰恰可以阻碍 HBV 对肝脏的感染! 我们知道 HBV 在我国的感染情况也是南高北低,那么南方人群中携带该突变(rs2296651,p.Ser267Phe)的人明显可以拥有更好的生存优势,只是需要在孕期付出较高胆汁酸水平的代价或者新生儿期出现黄疸,而这个代价对于生存在没有 HBV 疫苗年代的人们来说是可以接受的。
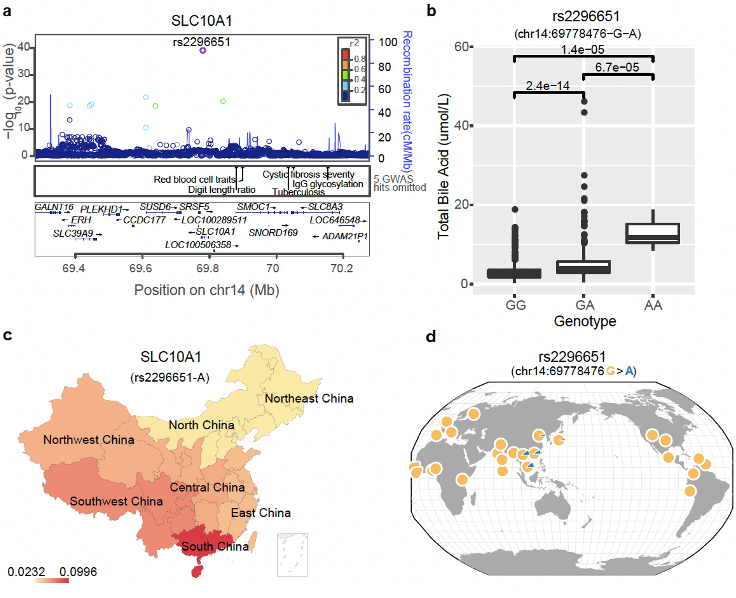
图 11. BIGCS 中 rs2296651 突变的频率分布
第二,我们意外发现脂质代谢可能存在年龄特异性的遗传效应(Age-specific genetic effect),也就是说,在不同的生命时期影响脂质代谢的主效基因可能是不同的。这个结果是在进行上述母子分组的 GWAS 分析过程中发现的。其实大家看上面 GWAS 的 Manhattan plot 也应该可以发现,对于同样的脂质代谢物,其显著的基因在母亲外周血和婴儿脐血中竟然是有差异的!例如在 SOAT2 基因座上,它在婴儿脐血中与低密度脂蛋白(LDL)水平相关,而在孕妇外周血中却与脂质水平无关;而与母体外周血中总胆固醇(TC)水平相关的 APOE 位点、与 LDL 水平相关的 CELSR2 位点以及与甘油三酯(TG)水平相关的 APOA5 位点在婴儿脐血中却没有表现出相似的遗传效应(图 12)。
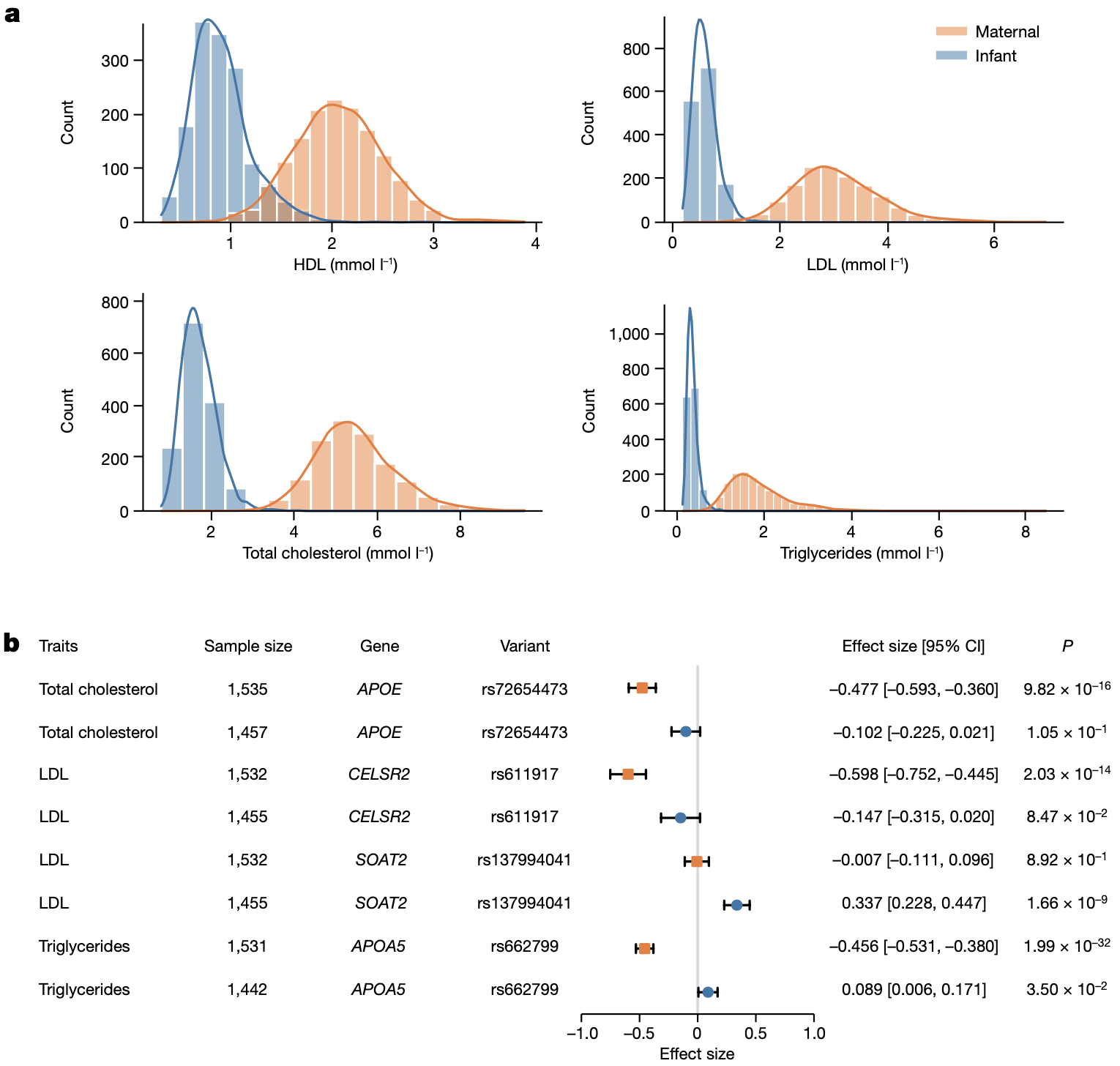
图 12. 脂代谢的年龄特异性遗传效应
值得一提的是这四个年龄特异的遗传效应位点,并不是靠肉眼看的,而是在关注到这个情况之后,做了两样本 t 检验(twosample-ttest) 把所有可能的位点过了一遍,最后才定位出来。而且我还分别对它们做了回归分析,才比较有把握地做出这个判断。
这是一个很有意思的发现。虽然我们这里只在脂质代谢中发现了这个情况,但它告诉我们同样的性状在不同的生命时期影响它的基因是有差异的,而且对于有些性状来说差异可能很大,这可以带来新的启发,比如是否有助于以后揭示衰老和发育相关的过程,也尤未可知。
另外,也意味着在人群中进行某些表型性状的 GWAS 时,你有必要按照不同生命时期对样本进行区分,否则可能会漏掉重要的信息。比如我们这里的这个发现至少可以为不同年龄阶段更准确地评估个体脂代谢相关疾病风险提供新的遗传学基础。
类似的现象如果可以在更多的人体性状中得到确认,还将意味着,通过一组既定的风险基因对不同年龄段的人进行表型/疾病风险预测的实际可行性和有效性将大大降低!
4. 跨代孟德尔随机化分析
如果说对于我们出生队列中哪些问题是最重要的,那么探索宫内环境的变化和遗传差异对子代健康的影响将是其中之一。而且胎儿在子宫中的生长发育同时也受其自身遗传特征的影响,如何剥离母体环境和遗传对子代的影响,从而准确地确定不同母体暴露对子代生长发育的因果关系一直都是一个困难的问题。如何才能解决或者部分解决这些问题,从而实现这个目标呢?在寻找解决方案的过程中,我们从辛辛那提儿童医院张戈老师的所发表的关于孟德尔随机化研究的文章中得到了很大的启发,我和刘斯洋教授还通过线上会议就所发表文章中的方法与张戈老师做了讨论。最后我们沿用了类似的思路设计并编写了适合我们队列的跨代孟德尔随机化方法。除了统计学上的一些假设之外,这里面有一个重要的点,就是要把母体中传递到子代的基因型(h1,maternal transmitted allele)、母体中非传递基因型(h2,maternal non-transmitted allele)和父亲的传递基因型(h3,paternal transmitted allele)区分出来(图 13)。
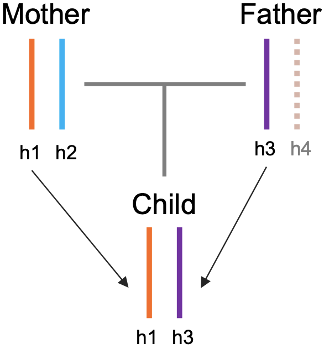
图 13. 亲代与子代基因型传递示意图
你可能也想到了,用 phasing 方法啊,确实是的,但这只对了一半。Phasing 出来的每一个 haplotype block 并非会严格按照相同的父母顺序进行排列,而是忽左忽右。这个也是在构造分析模型之前我们必须要先解决的一个问题,对于“父-母-子”这种 trio 家系来说,这个问题很简单,通过对比子代和父母的基因型差异就可以区分出来,但我们主要还是母-子这种双人组,这就不容易了。但好在每个phasing block 内的亲本顺序是固定了,那么就可以通过遍历其中可以实现三条染色体基因型区分的位点,从而将 h1,h2 和 h3 确定出来,然后再分别计算这三个单倍型的遗传得分(PRS) 最后将其作为工具变量实现暴露表型的跨代孟德尔随机化分析(图 14)。
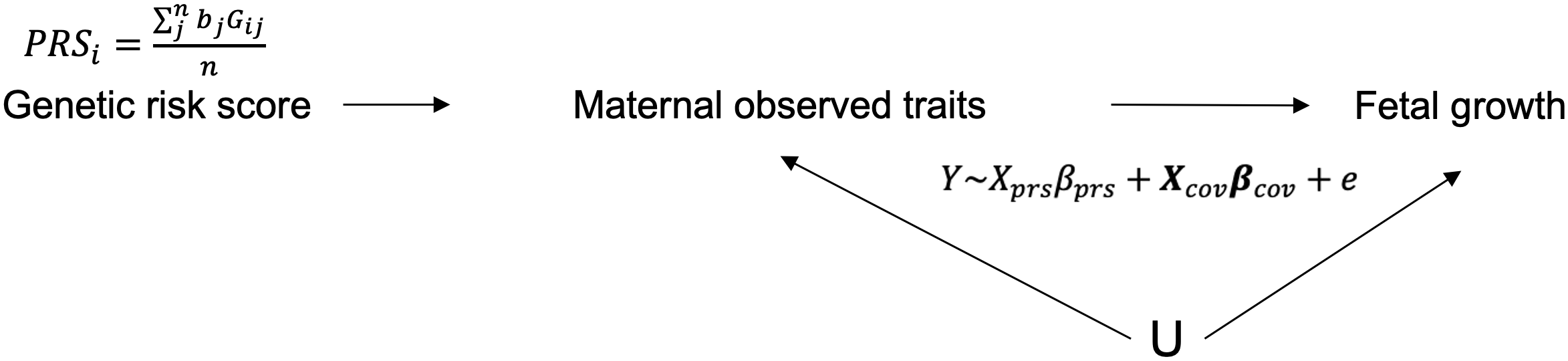
图 14. 孟德尔随机化分析框架图
但要注意,我们并不仅仅只是计算了上述三个单倍型的孟德尔随机化结果,还参考张戈老师的文章以及前面定相区分出的三个基因型结果,再更进一步地剥离宫内环境和遗传对胎儿生长发育的效应,建立了妊娠期暴露对胎儿生长的潜在因果关系的跨代孟德尔随机化方法。那我们是怎么做到的呢? 这里的细节很多,我只讲一个关键的地方。
回到上面图 13,这里只有 h2 可以较为纯粹地理解为它只直接影响了母体——即宫内环境(我们这里不考虑印记基因的影响),而 h1 虽然传递到子代,但它实际上依然在母体中,所以 h1 本身除了对子代有直接遗传影响外,理应还会对母体——宫内环境带来贡献,但贡献的程度应该是多少呢?我们这里做了两个简单的假设: (1)遗传和宫内环境对胎儿成长的影响是加性的(additive); (2)h1 对宫内环境的效应和 h2 程度相同,且 h1 对胎儿的遗传效应和 h3 相同。
在这两个假设前提下,我们就可以进行效应剥离了。比如母体总的宫内效应就应该是(β_h1+β_h2-β_h3)/2,而胎儿对自身的遗传效应则是 (β_h1+β_h3-β_h2)/2。
好了,模型构建出来之后可以正式进行数据分析了(实际上模型的构建过程并非是一蹴而就,而是做了很多次的尝试),主要结果我这里不打算再贴了,总的来说,除了和已知的一些发现相近之外,也有一些新的发现。比较有意思的发现是,在中国人群体中孕妇身高每增加 1cm,会增加 15g 婴儿的出生体重,而总胆汁酸每增加 1mmol/L,婴儿的出生身长则会减少 0.42 cm。另外,还有血压以及血糖也会对胎儿生长情况带来不同程度的影响(如下表)。
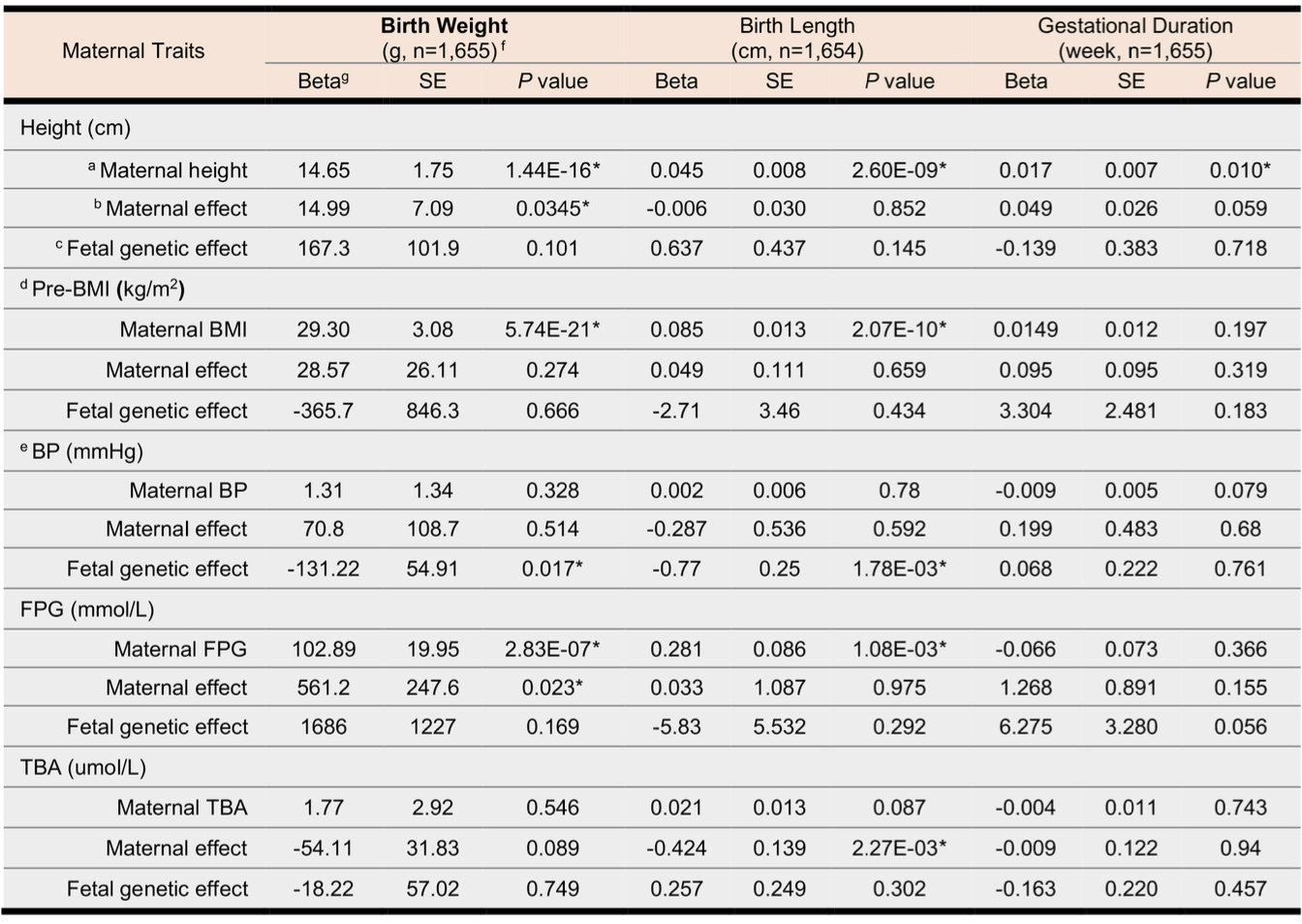
跨代孟德尔随机化分析的主要结果
以上,就是我们这个成果中的主要内容,以下是我们在项目执行过程中所开发的一些方法和流程:
1. 母婴遗传效应分离和 PRS 计算
2. 跨代孟德尔随机分析方法
3. 年龄特异遗传学效应分析方法
4. 快速全基因组测序数据分析流程:ilus
5. 突变数据系列质控方法
5. 基于 DNA 测序数据拷贝数差异的性别判断算法: rx_identifier 和 X/A ratio test
6. 完整的 GWAS Power 和 MR Power 分析程序
7. 出生队列基因组学网站 Genomics API tools:GDBIGtools (用于 GDBIG 网站)
8. 突变位点连锁不平衡程度计算程序:Pairwise LD calculation(用于 GDBIG 网站)
9. 基因填补流程(用于 GDBIG 网站)
10. GWAS 荟萃分析流程(用于 GDBIG 网站)
其他感想
科学的思路很重要,思路决定出路,不要放弃。在进行这项研究时,如果只是从群体基因组学或者群体遗传学方面进行突破,则我们已经不具备优势了。因为这个时候国内的 ChinaMAP、WBBC 和女娲(NyuWa)基因组都陆陆续续发表出来了,而且从样本量上来说我们也没有任何优势。我还记得当时我看到他们的文章的心情——焦虑又无奈,但你还是得往前走。我们一直咬住母子对的特点,一直咬住可以进行世代遗传学研究这一个优势,那只要我们能够在此处有所突破并构建好相关的方法,做别人所不能做的,如果最后还能有所发现,那么就还会有机会。
以上。
春节将至,提前祝愿大家身体好、学习好、工作好!龙年万事大吉,学运、财运样样昌隆!
原文链接
Huang, S. et al. The Born in Guangzhou Cohort Study enables generational genetic discoveries. Nature 626, 565-573 (2024).
Nature 评论:An early look at birth cohort genetics in China. https://www.nature.com/articles/d41586-024-00079-8
Nature 采访:New genetic variants found in large Chinese mother–baby study. https://www.nature.com/articles/d41586-024-00270-x
Claro Da Silva, T., et al. The solute carrier family 10 (SLC10): Beyond bile acid transport. Molecular Aspects of Medicine vol. 34 252–269 (2013).
Peng, L. et al. The p.Ser267Phe variant in SLC10A1 is associated with resistance to chronic hepatitis B. Hepatology 61, 1251–1260 (2015).
Liu, C. et al. The p.Ser267Phe variant of sodium taurocholate cotransporting polypeptide (NTCP) supports HBV infection with a low efficiency. Virology 522, 168–176 (2018).
该项目已通过伦理审查,符合国家法律法规及伦理规范,并严格保护参与者隐私及数据安全,并通过中国科技部人类遗传资源管理办公室的批准。
其他链接
- 国自然基金官网: 我国学者在宫内环境与遗传因素对子代健康影响的研究方面取得进展
- ChinaDaily 报道: Better ways to treat chronic diseases highlighted
- 广州出生队列公众号:《自然》重磅:广州出生队列揭示宫内环境和遗传差异对子代健康影响
- BioArt 公众号:Nature丨广州出生队列揭示母子健康传递的宫内秘境
- Nature 官方公众号:广州出生队列揭示宫内环境和遗传因素对子代健康的影响
基于大规模前瞻性出生队列的基因组研究,是人们理解遗传和环境对人体健康影响的重要方式,但这类研究在亚洲存在巨大空白。来自广州医科大学和中山大学的研究团队针对4053名研究对象开展了测序和分析。研究发现了东亚人群所特有的调控基因,这些基因与孕妇总胆汁酸、妊娠体重增加和婴儿脐血特征相关。此外,与脂质代谢相关的基因可能存在年龄特异性遗传效应。研究团队还采用跨代孟德尔随机化方法,剥离了宫内环境和遗传对胎儿生长发育的效应。这些发现为研究遗传、子宫内暴露和早期生活经历之间相互作用,及其塑造长期健康的机制奠定了基础。
本研究系亚洲首批调查母婴基因组与其健康之间关系的研究之一。
订阅
文章首发于我的个人公众号:helixminer(碱基矿工)
